農学生命情報科学特論I
|
|
|
- あおし あきます
- 7 years ago
- Views:
Transcription
 の hoge フォルダがデスクトップに残っているかもしれないのでご注意ください 農学生命情報科学 特論 I 第 3 回")
1 版 USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 前回 (6/23) の hoge フォルダがデスクトップに残っているかもしれないのでご注意ください 農学生命情報科学 特論 I 第 3 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected] 1
 Quality Control k-mer 解析 トリミング ( アダプター配列除去 ) 第 3 回 (2015 年 6 月 30 日 ) フィルタリング アセンブル マッピング カウント情報取得 教科書の 2.")
2 講義予定 第 1 回 (2015 年 6 月 16 日 ) NGS の普及により 以前は主にゲノム解析系で必要とされていた配列解析のためのスキルがトランスクリプトーム解析においても要求される時代になっています 本科目では 様々な局面で応用可能な配列解析系のスキルアップを目指し RNA-Seq に基づくトランスクリプトーム解析を題材とした講義を行います データベース データ取得 ファイル形式 Quality Control 教科書の 1.3 節周辺 第 2 回 (2015 年 6 月 23 日 ) Quality Control k-mer 解析 トリミング ( アダプター配列除去 ) 第 3 回 (2015 年 6 月 30 日 ) フィルタリング アセンブル マッピング カウント情報取得 教科書の 2.3 節周辺 第 4 回 (2015 年 7 月 7 日 ) クラスタリング 実験デザイン 分布 ( モデル ) 発現変動解析 教科書の 3.3 節周辺 教科書 2
3 Contents 先週の課題やエラーについて 課題 1 の解説 アダプター配列除去 (QuasR) 実行時のエラーは門田のミス Paired-end データの取り扱い フィルタリング (filtering) 基本形 : リード数が同じ paired-end の場合 (QuasR パッケージ ) 発展形 : リード数が異なる場合 (ShortRead パッケージ ) アセンブル (assemble) ゲノム用とトランスクリプトーム用 Rockhopper2( バクテリアのトランスクリプトーム用 ) マッピング (mapping) 基礎 オプション 仮想データ説明 マッピング本番 出力ファイル形式 オプションと結果の関係 カウント情報取得 実データ解析 QuasR でマッピング トリミング 高精度なアセンブルとマッピングへ 3

 の 55.")
4 k-mer 解析 上級 1 のコードをテンプレートにして Position 7 で特異的に出現している AAGAGCA を R で確認してみよう Head 関数出力結果は最初の 6 個 ( つまり position 1-6) までしか表示させていないので ここでは n=8 として position 1-8 まで表示させるようにしている たしかに position 7 で期待値 (Exp = 90.09) の 倍の出現回数になっていることが分かる 4


5 課題 1 上級 1 のコードをテンプレートにして 1 AGAGCAC 2GAGCACA 3 長さを含めて任意 の k-mer 解析を行い 得られた結果を示せ 1 5

6 課題 1 の解説 1AGAGCAC の k-mer 解析時にエラーが出たと思います この原因は AGAGCAC が 1 つも存在しないポジションがあった場合に そこが NA となるからです mean 関数実行時に要素中に NA が 1 つでもあると計算できないという罠があったのです 6
7 課題 1 の解説 Obs と打ち込んで 何が起こっているのかを悟る 画面に見えているだけでも AGAGCAC が存在しない position が少なくとも 2 ヶ所あることがわかる 7
 にするやり方 NA の要素を読み飛ばすオプションなので 平均値の計算が若干不正確になるが AGAGCAC が")
8 課題 1 の解説 最もシンプルな対処法は mean 関数中の na.rm オプションを TRUE ( デフォルトは FALSE) にするやり方 NA の要素を読み飛ばすオプションなので 平均値の計算が若干不正確になるが AGAGCAC が position 8 で相対的に高頻度に出現しているということを調べるという点では全く問題ない 茂木朋貴氏他数名提供情報 8
9 課題 1 の解説 平均値 ( この場合 ) さえ計算できれば Obs/Exp を計算可能 つまり プロファイルを描画することができる NA となっている場所が 2 ヶ所あることがわかる 9

10 課題 1 の解説 もっとよりよい方法は 1NA となった要素に 0 を入れること is.na 関数は 要素が NA のときに TRUE そうでないときに FALSE を返す 野間口達洋氏他数名提供情報 10

11 課題 1 の解説 0 代入前後で出現頻度の平均値が から に少し下がっているのがわかります 11

12 課題 1 の解説 こんな感じになります 2015 年 6 月 23 日の課題を該当部分のみ差し替えたいヒトは 回収箱に入れてかまいません 課題用紙は後ろにあります 先週提出分と突き合わせて評価しますので 考察部分など差し替える必要がない部分は空欄で構いません 1 12




13 バージョンアップ ひっそりと修正しています レポート中で問題点の指摘や解決法を示して頂いた方々に感謝 m( )m
14 Contents 先週の課題やエラーについて 課題 1 の解説 アダプター配列除去 (QuasR) 実行時のエラーは門田のミス Paired-end データの取り扱い フィルタリング (filtering) 基本形 : リード数が同じ paired-end の場合 (QuasR パッケージ ) 発展形 : リード数が異なる場合 (ShortRead パッケージ ) アセンブル (assemble) ゲノム用とトランスクリプトーム用 Rockhopper2( バクテリアのトランスクリプトーム用 ) マッピング (mapping) 基礎 オプション 仮想データ説明 マッピング本番 出力ファイル形式 オプションと結果の関係 カウント情報取得 実データ解析 QuasR でマッピング トリミング 高精度なアセンブルとマッピングへ 14

15 アダプター配列除去 1 param_nrec のところをいくら変えてもエラーが出続ける問題に多くの受講生が遭遇しました 理由はほぼ間違いなく 単純に私が preprocessreads 関数中に明示的に param_nrec を与えていなかったからです 結果として それが適切に反映されずにデフォルトの 1,000,000 がいつまでも使われていたということでしょう 2 中村浩正氏提供情報 15

16 アダプター配列除去 休み時間にでも動作確認してみてください きっとうまくいくことでしょう


17 Paired-end の取扱い 2015 年 6 月 23 日の講義資料
 では まだ pairedend データのアダプター配列除去には対応できていないようだといいましたが その後すぐに開発者が回避策")
18 Paired-end の取扱い QuasR (ver ) では まだ pairedend データのアダプター配列除去には対応できていないようだといいましたが その後すぐに開発者が回避策 (workaround) を教えてくれました Thanks to Dr. Stadler 18

19 回避策 基本戦略は single-end として別々にアダプター配列除去を行うが 出力ファイルのリード数が変わらないようにする です 乳酸菌 paired-end RNAseq データ (SRR626268) のアダプター配列除去を行う一連の手順の基本形は 例題 3-5 です 例題 3 では Forward 側のリードファイルを入力として FastQC でレポートされた TruSeq Adapter, Index 3 の除去を行っています 約 1 分 19


20 回避策 例題 4 では 例題 3 の出力ファイルを入力として もう 1 つレポートされていた TruSeq Adapter, Index 2 の除去を行っています FastQC 実行環境にない場合でも 用いた実験プロトコルが分かれば 候補となる adapter や primer 配列の一部で文字列検索すればある程度わかります もちろん この場合は Index 配列部分を含めたりしたほうがいいとは思います 約 1 分 20
")

21 回避策 例題 5 では Reverse 側のリードファイルを入力として FastQC でレポートされた Illumina Single End PCR Primer 1 の除去を行っています Primer 配列の場合は 逆相補鎖 ( reverse complement) を作成してから与えないとうまく除去できなかったことから このようにしています FastQC を実行できる環境にあれば overrepresented sequences 項目の possible source のところが No Hit になったかどうかでうまくいったかどうかの判定ができます 約 1 分 21
22 Contents 先週の課題やエラーについて 課題 1 の解説 アダプター配列除去 (QuasR) 実行時のエラーは門田のミス Paired-end データの取り扱い フィルタリング (filtering) 基本形 : リード数が同じ paired-end の場合 (QuasR パッケージ ) 発展形 : リード数が異なる場合 (ShortRead パッケージ ) アセンブル (assemble) ゲノム用とトランスクリプトーム用 Rockhopper2( バクテリアのトランスクリプトーム用 ) マッピング (mapping) 基礎 オプション 仮想データ説明 マッピング本番 出力ファイル形式 オプションと結果の関係 カウント情報取得 実データ解析 QuasR でマッピング トリミング 高精度なアセンブルとマッピングへ 22


23 フィルタリング : 基本形 リード数の揃った paired-end データを入力として 許容する N の数と最短配列長でフィルタリングするやり方を示します

24 フィルタリング : 基本形 この例題で用いているファイルは hoge4.fastq.gz と hoge5.fastq.gz でも構いません 中身は同じです 24

25 フィルタリング : 基本形 リード長が 18 塩基未満を捨て 1 塩基だけ N を許容するという閾値でフィルタリングをした結果 998,208 リードが残ったということ 約 2 分 25
26 フィルタリング : 発展形 基本戦略は paired-end を singleend として独立に取り扱い 最後に共通リードを抽出 例えば 1~3 あたりの任意のフィルタリングを singleend として独立に実行 最後に 4 でリード数の異なる paired-end のファイルを入力として 共通リードを出力 ここでは 1 の例題 6-8 を実行して得られたリード数の異なる paired-end ファイルを入力として 4 を実行するやり方を示します 個人見解としては 4 の入力ファイルの段階では FASTQ ではなく FASTA 形式でいいと思います 一連のフィルタリングでそこそこのクオリティが保証されているので わざわざクオリティ情報を保持しておく必要はないだろうという思想 26

27 フィルタリング : 発展形 1 参考 Small RNA-seq データのときはリードの右側にあるアダプター配列除去でしたが 乳酸菌 RNA-seq データのときは左側にアダプターがついているので Rpattern ではなく Lpattern になっている点に注意 2 27
28 フィルタリング : 発展形 参考 出力予定ファイルが存在する状態で実行するとエラーが出るので注意 28

29 フィルタリング : 発展形 参考 1 こんな感じになります 2 ここで見えている出力ファイルのサイズは 既に存在していた hoge5.fastq.gz のファイルサイズであり ここでのコピペ実行結果で得られたものではない点に注意

30 フィルタリング : 発展形 参考 hoge5.fastq.gz を hoge フォルダから削除して再度実行 30


31 フィルタリング : 発展形 参考 得られた hoge5.fastq.gz を入力として 例題 7 を実行 31

32 フィルタリング : 発展形 参考 例題 8 の paired-end reverse 側も実行 出力ファイルのサイズは 63,442,904 bytes 999,136 リード 32
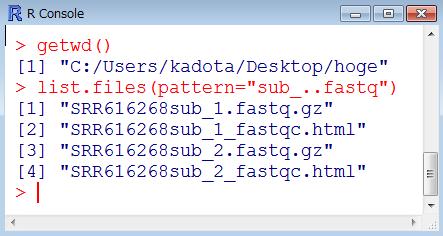
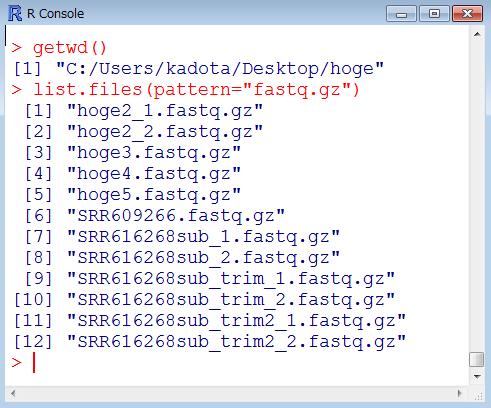
33 乳酸菌 RNA-seq データ 100 万リードのオリジナル?! ファイル SRR616268sub_1.fastq.gz:107 bp, 74,906,576 bytes SRR616268sub_2.fastq.gz:93 bp, 67,158,462 bytes 100 万リードのアダプター除去後のファイル SRR616268sub_trim2_1.fastq.gz(hoge4.fastq.gz): 71,343,605 bytes SRR616268sub_trim2_2.fastq.gz : 63,524,791 bytes 1 リード数の異なる paired-end のファイルを入力として 共通リードを出力するやり方を示します 107 bp 93 bp hoge2_1.fastq.gz: 998,208 リード 71,194,586 bytes hoge2_2.fastq.gz: 998,208 リード 63,375,473 bytes アダプター除去およびフィルタリング後のファイル SRR616268sub_trim_1.fastq.gz(hoge7.fastq.gz): 998,658 リード 71,227,695 bytes SRR616268sub_trim_2.fastq.gz(hoge8.fastq.gz): 999,136 リード 63,442,904 bytes 1 33
: 71,343,605 bytes SRR616268sub_trim2_2.fastq.gz : 63,524,791 bytes file.info 関数でもファイル名 サイズなど様々な情報をみることができます 107 bp 93 bp hoge2_1.fastq.gz: 998,208 リード 71,194,586 bytes hoge2_2.fastq.gz: 998,208 リード 63,375,473 bytes アダプター除去およびフィルタリング後のファイル SRR616268sub_trim_1.")
34 Tips:file.info 関数 100 万リードのオリジナル?! ファイル SRR616268sub_1.fastq.gz:107 bp, 74,906,576 bytes SRR616268sub_2.fastq.gz:93 bp, 67,158,462 bytes 100 万リードのアダプター除去後のファイル SRR616268sub_trim2_1.fastq.gz(hoge4.fastq.gz): 71,343,605 bytes SRR616268sub_trim2_2.fastq.gz : 63,524,791 bytes file.info 関数でもファイル名 サイズなど様々な情報をみることができます 107 bp 93 bp hoge2_1.fastq.gz: 998,208 リード 71,194,586 bytes hoge2_2.fastq.gz: 998,208 リード 63,375,473 bytes アダプター除去およびフィルタリング後のファイル SRR616268sub_trim_1.fastq.gz(hoge7.fastq.gz): 998,658 リード 71,227,695 bytes SRR616268sub_trim_2.fastq.gz(hoge8.fastq.gz): 999,136 リード 63,442,904 bytes 1 34


35 フィルタリング : 発展形 1 入力はリード数の異なる pairedend ファイル 出力はリード数が同じ paired-end ファイルです description 行の記述形式次第ではエラーが出ると思いますが 適宜変更してください 35
36 フィルタリング : 発展形 最初の 4 リード分の description 部分を表示 ここで見えているもので判断すると SRR は 2 つの入力ファイル中に存在する そして 少なくとも forward 側のファイル (*_1.fastq.gz) 中の 3 リード (SRR ) は reverse 側のファイル (*_2.fastq.gz) 中には存在しないことがわかる 36

37 フィルタリング : 発展形 共通リード抽出用の基本情報として リード ID の積集合 (intersection) を得るのが基本戦略 しかし description 部分の情報をそのまま使うと length=107 と length=93 という部分文字列の違いにより 1 同一リードを同一文字列として認識できないという問題がある 1 37
38 フィルタリング : 発展形 1 2 description 部分を眺め 部分文字列で同一リードを認識できる箇所を探す ここでは 1 description 部分を区切り文字 スペース ( ) で 分断し 21 番目の要素を取り出せば 赤下線部分のみの部分文字列で評価できるという戦略のもとでプログラムを組んでいる 38

39 フィルタリング : 発展形 Forward 側 うまくリード ID (SRR ) のみにできていることがわかります 39

40 フィルタリング : 発展形 Reverse 側 うまくリード ID (SRR ) のみにできていることがわかります 40
41 参考 Tips: コピペで使いまわす わざわざ別のオブジェクト名に置き換えているので 一見ややこしいですが 黒枠内のコードを使いまわせるので 間違いを減らせるというメリットがあります 41
 および reverse 側 (999,136) より少ないので妥当 42")
42 フィルタリング : 発展形 得られた共通リード数 (998,428) は forward 側 (998,658) および reverse 側 (999,136) より少ないので妥当 42

43 フィルタリング : 発展形 fastq1 オブジェクトの 1, 13, 17, 22 番目の要素が共通 43

44 フィルタリング : 発展形 is.element 関数は 右側の集合の中の要素が存在する位置を TRUE それ以外を FALSE として返す 共通である fastq1 オブジェクトの 1, 13, 17, 22 番目の要素が TRUE になっていることがわかる 44

45 フィルタリング : 発展形 共通リードのみなので リード数が揃っていることがわかる 45
46 乳酸菌 RNA-seq データ 100 万リードのオリジナル?! ファイル SRR616268sub_1.fastq.gz:107 bp, 74,906,576 bytes SRR616268sub_2.fastq.gz:93 bp, 67,158,462 bytes 100 万リードのアダプター除去後のファイル SRR616268sub_trim2_1.fastq.gz(hoge4.fastq.gz): 71,343,605 bytes SRR616268sub_trim2_2.fastq.gz : 63,524,791 bytes hoge2_1.fastq.gz: 998,208リード 71,194,586 bytes 2 hoge2_2.fastq.gz: 998,208リード 63,375,473 bytes アダプター除去およびフィルタリング後のファイル SRR616268sub_trim_1.fastq.gz(hoge7.fastq.gz): 998,658 リード 71,227,695 bytes SRR616268sub_trim_2.fastq.gz(hoge8.fastq.gz): 999,136 リード 63,442,904 bytes hoge1_1.fastq.gz: 998,428 リード 63,446,240 bytes hoge1_2.fastq.gz: 998,428 リード 56,019,410 bytes 1はデフォルトの 許容するN 数が2 最短配列長が14 のときの結果 2は若干厳しめの 許容するN 数が1 最短配列長が18 のときの結果なので 2のほうがリード数が若干少ない 107 bp 1 93 bp 46
47 Contents 先週の課題やエラーについて 課題 1 の解説 アダプター配列除去 (QuasR) 実行時のエラーは門田のミス Paired-end データの取り扱い フィルタリング (filtering) 基本形 : リード数が同じ paired-end の場合 (QuasR パッケージ ) 発展形 : リード数が異なる場合 (ShortRead パッケージ ) アセンブル (assemble) ゲノム用とトランスクリプトーム用 Rockhopper2( バクテリアのトランスクリプトーム用 ) マッピング (mapping) 基礎 オプション 仮想データ説明 マッピング本番 出力ファイル形式 オプションと結果の関係 カウント情報取得 実データ解析 QuasR でマッピング トリミング 高精度なアセンブルとマッピングへ 47



48 アセンブル : ゲノム用 1 ゲノム用 2 非モデル生物やヘテロ接合度の高い生物種用 3 微生物など小 ~ 中規模ゲノム配列決定用 アセンブルのアルゴリズム周辺は で web ページ内を検索

49 アセンブル : 転写物用 転写物 ( トランスクリプトーム ) 用


50 Rockhopper 1Windows のヒトは.exe ファイル Macintosh のヒトは.dmg ファイルで Rockhopper を起動 hoge - Rockhopper フォルダ中に Rockhopper_Results というフォルダがないヒトは ダブルクリックで起動したときに自動的に作成されます 3 最初 An error occurred while opening the file 的なポップアップが出現しますが 30 秒ほど待つと 4 Rockhopper の GUI が起動します

51 Rockhopper 1( 見づらいが )DE NOVO と赤字で書いている部分をクリック 2 入力ファイルを指定すべく BROWSE ボタンを押す


52 Rockhopper 1 ファイルの場所を指定 2Desktop 3hoge
53 乳酸菌 RNA-seq データ 100 万リードのオリジナル?! ファイル SRR616268sub_1.fastq.gz:107 bp, 74,906,576 bytes SRR616268sub_2.fastq.gz:93 bp, 67,158,462 bytes 100 万リードのアダプター除去後のファイル オリジナルの 100 万リードからなる生データファイルで実行 ( の発展課題と同じ ) 107 bp SRR616268sub_trim2_1.fastq.gz(hoge4.fastq.gz): 71,343,605 bytes SRR616268sub_trim2_2.fastq.gz : 63,524,791 bytes 93 bp hoge2_1.fastq.gz: 998,208 リード 71,194,586 bytes hoge2_2.fastq.gz: 998,208 リード 63,375,473 bytes アダプター除去およびフィルタリング後のファイル SRR616268sub_trim_1.fastq.gz(hoge7.fastq.gz): 998,658 リード 71,227,695 bytes SRR616268sub_trim_2.fastq.gz(hoge8.fastq.gz): 999,136 リード 63,442,904 bytes hoge1_1.fastq.gz: 998,428 リード 63,446,240 bytes hoge1_2.fastq.gz: 998,428 リード 56,019,410 bytes 53
54 Rockhopper:SE まずは single-end (SE) データとして実行 1 入力ファイルを選び 2 開く 3SUBMIT 約 1 分
55 Rockhopper:SE 1 入力ファイルは 1,000,000 リードだったので 一定の基準で内部的にフィルタリングしているのだろう 2 アセンブル後のリファレンス配列に対してマッピングした結果 1,964 リードがマップされたということだろう 3 アセンブルによって得られた転写物配列数は 8 個しかなかった 4 その平均配列長 (121 bp) と 5 中央値 (106 bp) 6 アセンブルされたトータルの塩基数は 974 これは 3 4 の結果 (8 121=968) とほぼ同じなので まあよしとしよう
56 乳酸菌 RNA-seq データ 100 万リードのオリジナル?! ファイル SRR616268sub_1.fastq.gz:107 bp, 74,906,576 bytes SRR616268sub_2.fastq.gz:93 bp, 67,158,462 bytes 100 万リードのアダプター除去後のファイル SRR616268sub_trim2_1.fastq.gz(hoge4.fastq.gz): 71,343,605 bytes SRR616268sub_trim2_2.fastq.gz : 63,524,791 bytes 次に reverse 側を single-end (SE) データとして de novo assemble 107 bp 93 bp hoge2_1.fastq.gz: 998,208 リード 71,194,586 bytes hoge2_2.fastq.gz: 998,208 リード 63,375,473 bytes アダプター除去およびフィルタリング後のファイル SRR616268sub_trim_1.fastq.gz(hoge7.fastq.gz): 998,658 リード 71,227,695 bytes SRR616268sub_trim_2.fastq.gz(hoge8.fastq.gz): 999,136 リード 63,442,904 bytes hoge1_1.fastq.gz: 998,428 リード 63,446,240 bytes hoge1_2.fastq.gz: 998,428 リード 56,019,410 bytes 56

57 Rockhopper:SE まずは single-end (SE) データとして実行 1 入力ファイルを選び 2 開く 3SUBMIT 約 1 分
 と 5 中央値 (228 bp) 6 アセンブルされたトータルの塩基数は 185,233 これは 3 4 の結果 (424 436=184,864) とほぼ同じなので まあよしとしよう 1 2")
58 Rockhopper:SE 1 入力ファイルは 1,000,000 リード 内部的にフィルタリングした結果 983,854 リードになったのだろう 2 アセンブル後のリファレンス配列に対してマッピングした結果 710,393 リードがマップされたということだろう 3 アセンブルによって得られた転写物配列数は 424 個 4 その平均配列長 (436 bp) と 5 中央値 (228 bp) 6 アセンブルされたトータルの塩基数は 185,233 これは 3 4 の結果 ( =184,864) とほぼ同じなので まあよしとしよう

59 Rockhopper:PE Paired-end データとしてアセンブル 1 念のため一旦消す 2 Options Clear cache というのがあったので こちらも念のためやってみる 数分フリーズ状態になりました 3Rockhopper アプリごと再起動しても キャッシュに残っているようなので Clear cache をやるのが無難かも


60 Rockhopper:PE しばらく経つと左のような画面になるので 1 paired-end の 1 つ目の forward 側のファイルを指定 2 双方向矢印 のところにカーソルをもっていくと Using paired-end reads; と書いてあるので ここをクリック 3OK を押す

61 Rockhopper:PE 1reverse 側のファイルを選択して 2 開く Paired-end の 2 つのファイルが見られるわけではないが 3SUBMIT


62 Rockhopper:PE 約 2 分で終わるが 残念ながらうまくアセンブルできていないようだ 後にこの原因は forward ファイル中の 3 側の adapter/primer 配列が主原因と判明 62

63 Rockhopper:PE それゆえ 1 Options - Parameter settings で 2Strand specific や Reverse complement reads のあたりをいじっても状況に変化はない
64 アセンブル結果まとめ いろいろやった結果のまとめです 64
65 Contents 先週の課題やエラーについて 課題 1 の解説 アダプター配列除去 (QuasR) 実行時のエラーは門田のミス Paired-end データの取り扱い フィルタリング (filtering) 基本形 : リード数が同じ paired-end の場合 (QuasR パッケージ ) 発展形 : リード数が異なる場合 (ShortRead パッケージ ) アセンブル (assemble) ゲノム用とトランスクリプトーム用 Rockhopper2( バクテリアのトランスクリプトーム用 ) マッピング (mapping) 基礎 オプション 仮想データ説明 マッピング本番 出力ファイル形式 オプションと結果の関係 カウント情報取得 実データ解析 QuasR でマッピング トリミング 高精度なアセンブルとマッピングへ 65

66 マッピング基礎 教科書 p81-89 基本イメージ マップされたリード数 = 発現量 ではないが マップされたリード数のカウント情報は 発現量推定の基本情報 基本的なマッピングプログラム (bowtie など ) を用いた場合 リファレンス配列 : ゲノム count T1 サンプルの RNA-Seq データ mapping 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 リファレンス配列 : トランスクリプトーム count 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 66
: AGG マッピング")
 リファレンス配列上のどの位置から転写されたものかという座標情報")
67 マッピング基礎 教科書 p81-89 マップされる側のリファレンス配列 :hoge4.fa マップする側のRNA-seqデータ ( リードと呼ばれる ): AGG マッピング = 大量高速文字列検索 という捉え方でよい マッピングプログラムの出力は ( どのリードが ) リファレンス配列上のどの位置から転写されたものかという座標情報 出力ファイル 67

 のおかげで Windows OS")
68 マッピング 1 教科書 p81-89 R パッケージとして提供されているものは圧倒的に少ないですが QuasR パッケージ (Gaidatzis et al., 2015) のおかげで Windows OS 上でもマッピングを気軽に利用できるようになりました これは内部的に Bowtie (Langmead et al., 2009) プログラムを利用しています 68
69 オプション QuasR パッケージは内部的に Bowtie を利用 マッピング時に多くのオプションを指定可能 -v : 許容するミスマッチ (mismatch) 数を指定するオプション -v 0 は リードがリファレンスに完全一致するもののみレポート -v 2 は 2 塩基ミスマッチまで許容してマップされうる場所を探索 -m : 出力するリード条件を指定するオプション -m 1 は 複数個所にマップされるリードを除外して 1 か所にのみマップされたリードをレポート -m 3 は 合計 3 か所にマップされるリードまでをレポート --best --strata : 最も少ないミスマッチ数でマップされるもののみ出力する という意思表示 これをつけずに -v 2 -m 1 などと指定すると たとえ完全一致 ( ミスマッチ数 0) で 1 か所にのみマップされるリードがあったとしても どこか別の場所で 1 塩基ミスマッチでマップされる個所があれば マップされうる場所が 2 か所ということを意味し そのリードは出力されなくなる それを防ぐのが主な目的... 教科書 p81-89 オプションは デフォルトである程度よきに計らってくれるが... 実際の挙動を完全に把握できる状況で様々なオプションを試したい 69
70 教科書 p81-89 仮想リファレンス配列 マップされる側のリファレンス配列 :ref_genome.fa chr3 と chr5 の違いは 2 番目と 7 番目の塩基のみ 主に -m オプション (-m 1:1 ヶ所にマップされるリードのみ出力 ) の違いの把握が可能 70

71 教科書 p81-89 仮想リファレンス配列 マップされる側のリファレンス配列 :ref_genome.fa コピペで作成しています 71
72 教科書 p81-89 仮想 RNA-seq リード マップする側の RNA-seq データ :sample_rnaseq1.fa 許容するミスマッチ数による違いや マップされるべき場所が完全に把握できるように リードの description 行に情報を記載 72

73 教科書 p81-89 仮想 RNA-seq リード マップする側の RNA-seq データ :sample_rnaseq1.fa コピペで作成しています 73
74 Contents 先週の課題やエラーについて 課題 1 の解説 アダプター配列除去 (QuasR) 実行時のエラーは門田のミス Paired-end データの取り扱い フィルタリング (filtering) 基本形 : リード数が同じ paired-end の場合 (QuasR パッケージ ) 発展形 : リード数が異なる場合 (ShortRead パッケージ ) アセンブル (assemble) ゲノム用とトランスクリプトーム用 Rockhopper2( バクテリアのトランスクリプトーム用 ) マッピング (mapping) 基礎 オプション 仮想データ説明 マッピング本番 出力ファイル形式 オプションと結果の関係 カウント情報取得 実データ解析 QuasR でマッピング トリミング 高精度なアセンブルとマッピングへ 74
75 マッピング本番 やってみましょう
76 マッピング本番 やってみましょう 1 2 複数の RNA-seq サンプルを実行できるようにリストファイルとして与える 許容するミスマッチ数は 0 個 ( -v 0 ) 1 か所にマップされるリードのみ出力 ( -m 1 ) 76

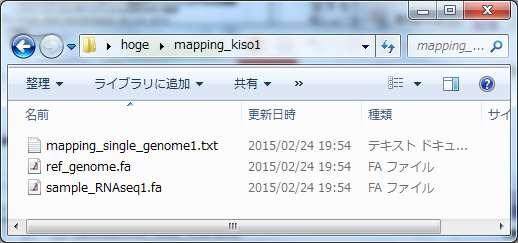
77 マッピング本番 hoge mapping_kiso1 に最低限必要な 3 つの入力ファイルが揃っていることを確認してコピペ ファイヤーフォールか何かでアクセス許可関連のポップアップが出たら OK を押す 77

78 出力ファイル形式 出力ファイルとして実際に取り扱うのは 1BAM 形式ファイルです 2 赤枠部分はランダムに発生させる文字列部分なので ヒトによって異なります
79 出力ファイル形式 実用上は BAM 形式 視覚上は BED 形式 ゲノム上のどの位置にどのリードがマッピングされたか ( トランスクリプトームの場合どの転写物配列上のどの位置にどのリードがマッピングされたか ) を表す出力ファイル形式は複数あります SAM (Sequence Alignment/Map) format SAMtools (Li et al., Bioinformatics, 25: , 2009) BAM (Binary Alignment/Map) format SAMtools (Li et al., Bioinformatics, 25: , 2009) BED (Browser Extensible Data) format... BEDtools (Quinlan et al., Bioinformatics, 26: , 2010) 79

80 出力ファイル形式 BED の最小限の情報は リード ID を含まない BAM 形式ファイル BED 形式ファイル 80
81 オプションと結果の関係 マップされなかったのは 赤枠の 8 リード中 3 リード -m 1 --best --strata -v 0 :0 ミスマッチで 1 か所にのみマップされるリードを出力 81
82 オプションと結果の関係 完全一致でも複数個所にマップされるために落とされたのは 2 リード -m 1: 1 ヶ所にマップされるリードのみ出力 -m 1 --best --strata -v 0 :0 ミスマッチで 1 か所にのみマップされるリードを出力 82
83 オプションと結果の関係 1 か所にのみマップされるがミスマッチのため落とされたのは 1 リード -v 0: 許容するミスマッチ数は 0 -m 1 --best --strata -v 0 :0 ミスマッチで 1 か所にのみマップされるリードを出力 83
84 Contents 先週の課題やエラーについて 課題 1 の解説 アダプター配列除去 (QuasR) 実行時のエラーは門田のミス Paired-end データの取り扱い フィルタリング (filtering) 基本形 : リード数が同じ paired-end の場合 (QuasR パッケージ ) 発展形 : リード数が異なる場合 (ShortRead パッケージ ) アセンブル (assemble) ゲノム用とトランスクリプトーム用 Rockhopper2( バクテリアのトランスクリプトーム用 ) マッピング (mapping) 基礎 オプション 仮想データ説明 マッピング本番 出力ファイル形式 オプションと結果の関係 カウント情報取得 実データ解析 QuasR でマッピング トリミング 高精度なアセンブルとマッピングへ 84

85 カウント情報取得 アノテーション情報を利用する場合 UCSC known Genes, Ensembl Genes など様々なテーブル名を指定可能 gene, exon, promoter, junction など様々なレベルを指定可能 アノテーション情報がない場合 R でのアノテーション情報利用は TranscriptDb が基本 マップされたリードの和集合領域を同定したのち 領域ごとのリード数をカウント BEDtools (Quinlan et al., 2010) 中の mergebed プログラムを実行して和集合領域同定後 intersectbed プログラムを実行してリード数をカウントする作業に相当 count 領域

86 カウント情報取得 アノテーション情報を利用する場合 UCSC known Genes, Ensembl Genes など様々なテーブル名を指定可能 gene, exon, promoter, junction など様々なレベルを指定可能 アノテーション情報がない場合 アノテーション情報がない場合の戦略は 複数サンプルの場合には領域が変わりうる Cufflinks を知っているヒトは cuffmerge と同じイメージだと思えばよい マップされたリードの和集合領域を同定したのち 領域ごとのリード数をカウント BEDtools (Quinlan et al., 2010) 中の mergebed プログラムを実行して和集合領域同定後 intersectbed プログラムを実行してリード数をカウントする作業に相当 sample1 count sample2 86


87 カウント情報取得 1 アノテーション情報がない場合のやり方です


88 カウント情報取得 1 hoge mapping_kiso1 に入力ファイルは揃っています マッピング結果ファイルが既に存在しますが log ファイルを見て同じオプションで実行した結果があるかどうかを自動判定します 再度マッピングを実行する必要がない場合は 以前に得られていた bam ファイルを入力として 速やかに 1 のところの作業に移行します 1 88


89 カウント情報取得 1 出力ファイルは何も指定していませんが *_range.txt という名前のファイルが作成されます これは.bam という名前のファイルを内部的に入力として読み込み その文字列中の.bam を _range.txt に置換したものを出力ファイル名として自動作成しているからそうなります 89

90 カウント情報取得 1.bed ファイルと *_range.txt ファイルを見比べると理解が深まるでしょう *_range.txt ファイルの一番右側の列がカウント情報です *.bed *_range.txt 90



91 カウント情報取得 2 複数の RNA-seq リードファイルのマッピングからカウントデータ取得までを一気に行う例です

92 カウント情報取得 2 hoge mapping_kiso2 に入力ファイルは揃っています 92

93 カウント情報取得 2 コピペ 出力ファイル群のうち 主に取り扱うのは カウントデータを含むファイル (hoge4_count.txt) 93
")
94 カウント情報取得 2 リストファイル中で指定したサンプル名 (sample1 と sample2) がカウントデータ行列の列名となります 94
95 Contents 先週の課題やエラーについて 課題 1 の解説 アダプター配列除去 (QuasR) 実行時のエラーは門田のミス Paired-end データの取り扱い フィルタリング (filtering) 基本形 : リード数が同じ paired-end の場合 (QuasR パッケージ ) 発展形 : リード数が異なる場合 (ShortRead パッケージ ) アセンブル (assemble) ゲノム用とトランスクリプトーム用 Rockhopper2( バクテリアのトランスクリプトーム用 ) マッピング (mapping) 基礎 オプション 仮想データ説明 マッピング本番 出力ファイル形式 オプションと結果の関係 カウント情報取得 実データ解析 QuasR でマッピング トリミング 高精度なアセンブルとマッピングへ 95


96 実データ解析 アノテーション情報がない場合のやり方です 1 96

97 実データ解析 hoge mapping_paired1 に解析したい fastq.gz ファイルを移動させて実行 約 8 分 97

98 実データ解析 1 このあたりで約 1 分 リファレンス配列のインデックス作成部分 約 3MB の乳酸菌ゲノムなので短時間で終わる ヒトゲノムだと数十分から数時間といったところか 2 のところで約 7 分 オリジナルの総リード数は 1.4 億 1/100 以下の 100 万リードだから 10 分足らずで終わる

99 実データ解析 1 マッピングの実体である qalign 関数実行前後の時間を計測し /60 = 7.05 分という結果を得ている 1 99
 3 マッピング結果の概観 forward 側と reverse 側合わせて 200 万リード中 8,204 リードしかマップされてない!")
100 実データ解析 1 マッピングに用いられたオプション ( パラメータ ) 情報 2 これがメインのマッピング結果ファイルの名前 ( いわゆる BAM ファイル ) 3 マッピング結果の概観 forward 側と reverse 側合わせて 200 万リード中 8,204 リードしかマップされてない!

101 実データ解析 1list.files を用いて ここまでの作業で作成されたファイルを概観 元は黒枠の 4 つのファイルのみだったが マッピングが終わった時点で数多くのファイルが作成されていることが分かる 2 マッピング結果を取扱うときの基本は BAM ファイル 3 マッピングに用いられたオプション ( パラメータ ) 情報は この log ファイルからも閲覧可能

102 QC レポート作成 1sub 関数を用いて黒下線で見られるファイル名中の拡張子部分.bam を _QC.pdf に置換したファイル名を作成して out_f に格納している 拡張子部分周辺の文字列だけを変更する程度の出力ファイル名であれば このように自動的に作成することができる 数十秒程度 1 102
 2 得られた BED 形式ファイルの中身 マップされたリード数が 8,204 個であったことから BED ファイルの行数が")
103 BED ファイル作成 1 これは BAM ファイルがバイナリ形式でそんなものだと慣れてしまえば不必要 単純に視覚的に見やすいように bam ファイルを読み込んで BED 形式ファイルを作成しているところ ( つまりファイル形式の変換 ) 2 得られた BED 形式ファイルの中身 マップされたリード数が 8,204 個であったことから BED ファイルの行数が 8,204 行なのは妥当


104 QC レポート概観 1QC レポートを眺める 最初のページは quality score 分布 2forward 側と 3 reverse 側で左右に表示されている カイコ small RNA-seq データ (single-end) で左側のみにしか表示されていなかったときは違和感を覚えたが paired-end データで眺めると至極妥当な配置であったことが分かる
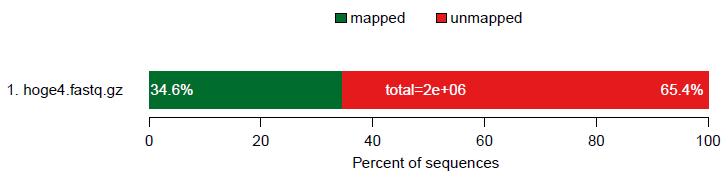
 4 4page 目 リード全体の mapped/unmapped の割合を表示 確かに 200 万リード中 8,204")
105 QC レポート概観 2 3 1QC レポートの 2page 目 2Forward のリードの左側は確かにガタガタしていたので妥当だが 3 右側もこんなになってたっけ?! と思いつつ ( 後にこれが犯人と判明 ) 4 4page 目 リード全体の mapped/unmapped の割合を表示 確かに 200 万リード中 8,204 リードしかマップされてなかったので妥当

106 QC レポート概観 15page 目 200 万リード中 8,204 リードマップされていたが paired-end なので ここでは分母を 8,204/2 = 4, e+03 と表現しているようだ 1 ヶ所にのみマップされたのは全体の 91.7% 26page 目 これはおそらくマップされたリードの中でどのポジションにミスマッチがあったかを示す部分 3 と 4 を眺めて 確かに FastQC で眺めていたのは最初の 50 bp 分で adapters/primers の除去は 5 側のみだった!
107 アセンブル結果再考 このぶざまな状況を打破する糸口が完璧に見つかった! 107
108 Contents 先週の課題やエラーについて 課題 1 の解説 アダプター配列除去 (QuasR) 実行時のエラーは門田のミス Paired-end データの取り扱い フィルタリング (filtering) 基本形 : リード数が同じ paired-end の場合 (QuasR パッケージ ) 発展形 : リード数が異なる場合 (ShortRead パッケージ ) アセンブル (assemble) ゲノム用とトランスクリプトーム用 Rockhopper2( バクテリアのトランスクリプトーム用 ) マッピング (mapping) 基礎 オプション 仮想データ説明 マッピング本番 出力ファイル形式 オプションと結果の関係 カウント情報取得 実データ解析 QuasR でマッピング トリミング 高精度なアセンブルとマッピングへ 108
 考えられる戦略を述べよ 課題 3: 今回の結果を踏まえ リファレンスゲノムがない ( マッピング結果がない ) 場合の高精度なアセンブル戦略について述べよ")
109 課題 1, 2, 3 課題 1: Rockhopper のアセンブル結果 を QuasR マッピング結果の QC レポート と絡めて説明せよ 課題 2: どうすればマッピング結果を改善できるのか ( マップ率を上げることができるのか ) 考えられる戦略を述べよ 課題 3: 今回の結果を踏まえ リファレンスゲノムがない ( マッピング結果がない ) 場合の高精度なアセンブル戦略について述べよ 109


110 課題 4 課題 4:1 は FastQC の Per base sequence content 2 は QuasR の QC レポートファイル中の同様な結果である 赤点線枠部分に違いが見られるが この理由について主に描画方法の観点から考察せよ よく分からないヒトは別の観点でもよい もちろん色の違いは本質的ではない
111 Rockhopper リトライ これだけ結果が変わります かなりイケてます 111

112 QuasR リトライ これだけ結果が変わります かなりイケてます 112
基本的な利用法
 (R で ) 塩基配列解析 基本的な利用法 Macintosh 版 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける 1. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール 2. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握
(R で ) 塩基配列解析 基本的な利用法 Macintosh 版 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける 1. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール 2. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握
Rインストール手順
 R 本体は最新のリリース版 R パッケージは 必要最小限プラスアルファ の推奨インストール手順を示します R 本体とパッケージのインストール Windows 版 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected] http://www.iu.a.u-tokyo.ac.jp/~kadota/
R 本体は最新のリリース版 R パッケージは 必要最小限プラスアルファ の推奨インストール手順を示します R 本体とパッケージのインストール Windows 版 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected] http://www.iu.a.u-tokyo.ac.jp/~kadota/
特論I
 2016.02.01 版 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 3 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 [email protected] Jun 25, 2014 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG
2016.02.01 版 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 3 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 [email protected] Jun 25, 2014 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG
NGSハンズオン講習会
 207.08.08 版 プラスアルファの内容です NGS 解析 ( 初 ~ 中級 ) ゲノムアセンブリ後の各種解析の補足資料 ( プラスアルファ ) 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム寺田朋子 門田幸二 Aug 29-30 207 Contents Gepard でドットプロット 連載第 8 回 W5-3 で最も長い sequence 同士のドットプロットを実行できなかったが
207.08.08 版 プラスアルファの内容です NGS 解析 ( 初 ~ 中級 ) ゲノムアセンブリ後の各種解析の補足資料 ( プラスアルファ ) 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム寺田朋子 門田幸二 Aug 29-30 207 Contents Gepard でドットプロット 連載第 8 回 W5-3 で最も長い sequence 同士のドットプロットを実行できなかったが
NGSデータ解析入門Webセミナー
 NGS データ解析入門 Web セミナー : RNA-Seq 解析編 1 RNA-Seq データ解析の手順 遺伝子発現量測定 シークエンス マッピング サンプル間比較 機能解析など 2 CLC Genomics Workbench 使用ツール シークエンスデータ メタデータのインポート NGS data import Import Metadata クオリティチェック Create Sequencing
NGS データ解析入門 Web セミナー : RNA-Seq 解析編 1 RNA-Seq データ解析の手順 遺伝子発現量測定 シークエンス マッピング サンプル間比較 機能解析など 2 CLC Genomics Workbench 使用ツール シークエンスデータ メタデータのインポート NGS data import Import Metadata クオリティチェック Create Sequencing
NGS速習コース
 バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-3. R 各種パッケージ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) [email protected] http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-3. R 各種パッケージ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) [email protected] http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
シーケンサー利用技術講習会 第10回 サンプルQC、RNAseqライブラリー作製/データ解析実習講習会
 シーケンサー利用技術講習会 第 10 回サンプル QC RNAseq ライブ ラリー作製 / データ解析実習講習会 理化学研究所ライフサイエンス技術基盤研究センターゲノムネットワーク解析支援施設田上道平 次世代シーケンサー Sequencer File Format Output(Max) Read length Illumina Hiseq2500 Fastq 600 Gb 100 bp Life
シーケンサー利用技術講習会 第 10 回サンプル QC RNAseq ライブ ラリー作製 / データ解析実習講習会 理化学研究所ライフサイエンス技術基盤研究センターゲノムネットワーク解析支援施設田上道平 次世代シーケンサー Sequencer File Format Output(Max) Read length Illumina Hiseq2500 Fastq 600 Gb 100 bp Life
基本的な利用法
 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握 ) 4. ありがちなミス のところで示しているエラーメッセージとその原因をきっちり理解
到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握 ) 4. ありがちなミス のところで示しているエラーメッセージとその原因をきっちり理解
リード・ゲノム・アノテーションインポート
 リード ゲノム アノテーションインポート 1 Location と Folder ロケーション フォルダ Genomics Workbenchではデータを以下のような階層構造で保存可能です フォルダの一番上位の階層を Location と呼び その下の階層を Folder と呼びます データの保存場所はロケーション毎に設定可能です たとえばあるデータは C ドライブに保存し あるデータは D ドライブに保存するといった事が可能です
リード ゲノム アノテーションインポート 1 Location と Folder ロケーション フォルダ Genomics Workbenchではデータを以下のような階層構造で保存可能です フォルダの一番上位の階層を Location と呼び その下の階層を Folder と呼びます データの保存場所はロケーション毎に設定可能です たとえばあるデータは C ドライブに保存し あるデータは D ドライブに保存するといった事が可能です
PowerPoint プレゼンテーション
 V1 次世代シークエンサ実習 II 本講義の内容 Reseq 解析 RNA-seq 解析 公開データ取得 クオリティコントロール マッピング 変異検出 公開データ取得 クオリティコントロール マッピング 発現定量 FPKM を算出します 2 R N A - s e q とは メッセンジャー RNA(mRNA) をキャプチャして次世代シーケンサーでシーケンシングする手法 リファレンスがある生物種の場合
V1 次世代シークエンサ実習 II 本講義の内容 Reseq 解析 RNA-seq 解析 公開データ取得 クオリティコントロール マッピング 変異検出 公開データ取得 クオリティコントロール マッピング 発現定量 FPKM を算出します 2 R N A - s e q とは メッセンジャー RNA(mRNA) をキャプチャして次世代シーケンサーでシーケンシングする手法 リファレンスがある生物種の場合
ChIP-seq
 ChIP-seq 1 ChIP-seq 解析原理 ChIP サンプルのフラグメントでは タンパク質結合部位付近にそれぞれ Forward と Reverse のリードがマップされることが予想される ChIP のサンプルでは Forward と Reverse のリードを 3 側へシフトさせ ChIP のピークを算出する コントロールサンプルでは ChIP のサンプルとは異なり 特定の場所に多くマップされないため
ChIP-seq 1 ChIP-seq 解析原理 ChIP サンプルのフラグメントでは タンパク質結合部位付近にそれぞれ Forward と Reverse のリードがマップされることが予想される ChIP のサンプルでは Forward と Reverse のリードを 3 側へシフトさせ ChIP のピークを算出する コントロールサンプルでは ChIP のサンプルとは異なり 特定の場所に多くマップされないため
特論I
 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 2 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 [email protected] 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG 概論 現状や展望など 講義のみ 第 2 回 (2014
講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 2 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 [email protected] 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG 概論 現状や展望など 講義のみ 第 2 回 (2014
PrimerArray® Analysis Tool Ver.2.2
 研究用 PrimerArray Analysis Tool Ver.2.2 説明書 v201801 PrimerArray Analysis Tool Ver.2.2 は PrimerArray( 製品コード PH001 ~ PH007 PH009 ~ PH015 PN001 ~ PN015) で得られたデータを解析するためのツールで コントロールサンプルと 1 種類の未知サンプル間の比較が可能です
研究用 PrimerArray Analysis Tool Ver.2.2 説明書 v201801 PrimerArray Analysis Tool Ver.2.2 は PrimerArray( 製品コード PH001 ~ PH007 PH009 ~ PH015 PN001 ~ PN015) で得られたデータを解析するためのツールで コントロールサンプルと 1 種類の未知サンプル間の比較が可能です
GWB
 NGS データ解析入門 Web セミナー : De Novo シークエンス解析編 1 NGS 新規ゲノム配列解析の手順 シークエンス 遺伝子領域の検出 アセンブル データベース検索 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 前処理 コンティグ配列の作成 CLC Genomics Workbench 遺伝子領域の検出 Blast2GO PRO データベース検索
NGS データ解析入門 Web セミナー : De Novo シークエンス解析編 1 NGS 新規ゲノム配列解析の手順 シークエンス 遺伝子領域の検出 アセンブル データベース検索 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 前処理 コンティグ配列の作成 CLC Genomics Workbench 遺伝子領域の検出 Blast2GO PRO データベース検索
ゲノム情報解析基礎 ~ Rで塩基配列解析 ~
 ネット接続できないヒトも ダブルクリックでローカルに r_seq.html を起動可能です 実習は デスクトップ上にある hoge フォルダの中身が以下の状態を想定して行います (R で ) 塩基配列解析の利用法 : GC 含量計算から発現変動解析まで東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected]
ネット接続できないヒトも ダブルクリックでローカルに r_seq.html を起動可能です 実習は デスクトップ上にある hoge フォルダの中身が以下の状態を想定して行います (R で ) 塩基配列解析の利用法 : GC 含量計算から発現変動解析まで東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected]
スライド 1
 ラベル屋さん HOME かんたんマニュアル リンクコース 目次 STEP 1-2 : ( 基礎編 ) 用紙の選択と文字の入力 STEP 3 : ( 基礎編 ) リンクの設定 STEP 4 : ( 基礎編 ) リンクデータの入力と印刷 STEP 5 : ( 応用編 ) リンクデータの入力 1 STEP 6 : ( 応用編 ) リンクデータの入力 2 STEP 7-8 : ( 応用編 ) リンク機能で使ったデータをコピーしたい場合
ラベル屋さん HOME かんたんマニュアル リンクコース 目次 STEP 1-2 : ( 基礎編 ) 用紙の選択と文字の入力 STEP 3 : ( 基礎編 ) リンクの設定 STEP 4 : ( 基礎編 ) リンクデータの入力と印刷 STEP 5 : ( 応用編 ) リンクデータの入力 1 STEP 6 : ( 応用編 ) リンクデータの入力 2 STEP 7-8 : ( 応用編 ) リンク機能で使ったデータをコピーしたい場合
機能ゲノム学
 08.05.08 版 講義資料 PDF が講義のページからダウンロード可能です 講義資料の印刷物はありません 課題用の A4 一枚はあります 第 回出席予定の持込み PC の方は 当日までに Java のインストールをしておいてください 機能ゲノム学第 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム 微生物科学イノベーション連携研究機構門田幸二 ( かどたこうじ ) [email protected]
08.05.08 版 講義資料 PDF が講義のページからダウンロード可能です 講義資料の印刷物はありません 課題用の A4 一枚はあります 第 回出席予定の持込み PC の方は 当日までに Java のインストールをしておいてください 機能ゲノム学第 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム 微生物科学イノベーション連携研究機構門田幸二 ( かどたこうじ ) [email protected]
GWB
 NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 CLC Genomics Workbench 使用ツール シークエンスデータのインポート NGS data import クオリティチェック QC for Sequencing Reads Trim Reads 参照ゲノム配列へのマッピング 再アライメント
NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 CLC Genomics Workbench 使用ツール シークエンスデータのインポート NGS data import クオリティチェック QC for Sequencing Reads Trim Reads 参照ゲノム配列へのマッピング 再アライメント
Si 知識情報処理
 242311 Si, 285301 MS 第 12 回 竹平真則 [email protected] 2015/12/21 1 本日の内容 1. 先週のおさらい 2. PHP のスクリプトを実際に動かしてみる 3. RDB についての説明 2015/12/21 2 資料の URL http://peacenet.info/m2is 2015/12/21 3 注意事項 ( その
242311 Si, 285301 MS 第 12 回 竹平真則 [email protected] 2015/12/21 1 本日の内容 1. 先週のおさらい 2. PHP のスクリプトを実際に動かしてみる 3. RDB についての説明 2015/12/21 2 資料の URL http://peacenet.info/m2is 2015/12/21 3 注意事項 ( その
GWB_RNA-Seq_
 CLC Genomics Workbench ウェブトレーニングセミナー : RNA-Seq 編 フィルジェン株式会社バイオサイエンス部 ([email protected]) 1 Advanced RNA-Seq プラグイン CLC Genomics Workbench 9.0 / Biomedical Genomics Workbench 3.0 以降で使用可能な無償プラグイン RNA-Seq
CLC Genomics Workbench ウェブトレーニングセミナー : RNA-Seq 編 フィルジェン株式会社バイオサイエンス部 ([email protected]) 1 Advanced RNA-Seq プラグイン CLC Genomics Workbench 9.0 / Biomedical Genomics Workbench 3.0 以降で使用可能な無償プラグイン RNA-Seq
Rでゲノム・トランスクリプトーム解析
 R でゲノム トランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 自己紹介 1995 年 3 月 高知工業高等専門学校 工業化学科卒業 1997 年 3 月 東京農工大学 工学部 物質生物工学科卒業
R でゲノム トランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 自己紹介 1995 年 3 月 高知工業高等専門学校 工業化学科卒業 1997 年 3 月 東京農工大学 工学部 物質生物工学科卒業
CubePDF ユーザーズマニュアル
 CubePDF ユーザーズマニュアル 2018.11.22 第 13 版 1 1. PDF への変換手順 CubePDF は仮想プリンターとしてインストールされます そのため Web ブラウザや Microsoft Word, Excel, PowerPoint など印刷ボタンのあるアプリケーションであればどれでも 次の 3 ステップで PDF へ変換することができます 1. PDF 化したいものを適当なアプリケーションで表示し
CubePDF ユーザーズマニュアル 2018.11.22 第 13 版 1 1. PDF への変換手順 CubePDF は仮想プリンターとしてインストールされます そのため Web ブラウザや Microsoft Word, Excel, PowerPoint など印刷ボタンのあるアプリケーションであればどれでも 次の 3 ステップで PDF へ変換することができます 1. PDF 化したいものを適当なアプリケーションで表示し
Microsoft Word - Word1.doc
 Word 2007 について ( その 1) 新しくなった Word 2007 の操作法について 従来の Word との相違点を教科書に沿って説明する ただし 私自身 まだ Word 2007 を使い込んではおらず 間違いなどもあるかも知れない そうした点についてはご指摘いただければ幸いである なお 以下において [ ] で囲った部分は教科書のページを意味する Word の起動 [p.47] Word
Word 2007 について ( その 1) 新しくなった Word 2007 の操作法について 従来の Word との相違点を教科書に沿って説明する ただし 私自身 まだ Word 2007 を使い込んではおらず 間違いなどもあるかも知れない そうした点についてはご指摘いただければ幸いである なお 以下において [ ] で囲った部分は教科書のページを意味する Word の起動 [p.47] Word
PowerPoint Presentation
 エピジェノミクス解析編 2016/08/10 Filgen ChIP-seq (Transfactor & Histone), Bisulfite webex seminar 株式会社キアゲンアプライドアドバンストゲノミクス宮本真理, PhD 1 アジェンダ ChIP-seq 解析 Transcription Factor ChIP-seq Histone ChIP-seq Bisulfite-seq
エピジェノミクス解析編 2016/08/10 Filgen ChIP-seq (Transfactor & Histone), Bisulfite webex seminar 株式会社キアゲンアプライドアドバンストゲノミクス宮本真理, PhD 1 アジェンダ ChIP-seq 解析 Transcription Factor ChIP-seq Histone ChIP-seq Bisulfite-seq
Rでゲノム・トランスクリプトーム解析
 06.03.05 版 実習用 PC のデスクトップ上に hoge フォルダがあります この中に解析に必要な入力ファイルがあります ネットワーク不具合時は ローカル環境で html ファイルを起動して各自対応してください R で塩基配列解析 : ゲノム解析からトランスクリプトーム解析まで 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ )
06.03.05 版 実習用 PC のデスクトップ上に hoge フォルダがあります この中に解析に必要な入力ファイルがあります ネットワーク不具合時は ローカル環境で html ファイルを起動して各自対応してください R で塩基配列解析 : ゲノム解析からトランスクリプトーム解析まで 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ )
( 目次 ) 1. はじめに 開発環境の準備 仮想ディレクトリーの作成 ASP.NET のWeb アプリケーション開発環境準備 データベースの作成 データベースの追加 テーブルの作成
 1. はじめに 開発環境の準備 仮想ディレクトリーの作成 ASP.NET のWeb アプリケーション開発環境準備 データベースの作成 データベースの追加 テーブルの作成") KDDI ホスティングサービス (G120, G200) ブック ASP.NET 利用ガイド ( ご参考資料 ) rev.1.0 KDDI 株式会社 1 ( 目次 ) 1. はじめに... 3 2. 開発環境の準備... 3 2.1 仮想ディレクトリーの作成... 3 2.2 ASP.NET のWeb アプリケーション開発環境準備... 7 3. データベースの作成...10 3.1 データベースの追加...10
KDDI ホスティングサービス (G120, G200) ブック ASP.NET 利用ガイド ( ご参考資料 ) rev.1.0 KDDI 株式会社 1 ( 目次 ) 1. はじめに... 3 2. 開発環境の準備... 3 2.1 仮想ディレクトリーの作成... 3 2.2 ASP.NET のWeb アプリケーション開発環境準備... 7 3. データベースの作成...10 3.1 データベースの追加...10
機能ゲノム学(第6回)
") RNA-Seqデータ解析における正規化法の選択 :RPKM 値でサンプル間比較は危険?! 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 よりよい正規化法とは? その正規化法によって得られたデータを用いて発現変動の度合いでランキングしたときに
RNA-Seqデータ解析における正規化法の選択 :RPKM 値でサンプル間比較は危険?! 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 よりよい正規化法とは? その正規化法によって得られたデータを用いて発現変動の度合いでランキングしたときに
機能ゲノム学(第6回)
") RNA-Seq データ解析リテラシー 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 2009 年ごろの私 次世代シーケンサー (NGS) 解析についての認識 単に短い塩基配列が沢山あるだけでしょ 得られる配列データって
RNA-Seq データ解析リテラシー 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 2009 年ごろの私 次世代シーケンサー (NGS) 解析についての認識 単に短い塩基配列が沢山あるだけでしょ 得られる配列データって
1. MEGA 5 をインストールする 1.1 ダウンロード手順 MEGA のホームページ (http://www.megasoftware.net/index.php) から MEGA 5 software をコンピュータにインストールする 2. 塩基配列を決定する 2.1 Alignment E
 から MEGA 5 software をコンピュータにインストールする 2. 塩基配列を決定する 2.1 Alignment E") MEGA 5 を用いた塩基配列解析法および分子系統樹作成法 Ver.1 Update: 2012.04.01 ウイルス 疫学研究領域井関博 < 内容 > 1. MEGA 5 をインストールする 1.1 ダウンロード手順 2. 塩基配列を決定する 2.1 Alignment Explorer の起動 2.2 シークエンスデータの入力 2.2.1 テキストファイルから読み込む場合 2.2.2 波形データから読み込む場合
MEGA 5 を用いた塩基配列解析法および分子系統樹作成法 Ver.1 Update: 2012.04.01 ウイルス 疫学研究領域井関博 < 内容 > 1. MEGA 5 をインストールする 1.1 ダウンロード手順 2. 塩基配列を決定する 2.1 Alignment Explorer の起動 2.2 シークエンスデータの入力 2.2.1 テキストファイルから読み込む場合 2.2.2 波形データから読み込む場合
<4D F736F F D F82C A815B835982B782E98FEA8D8782CC91CE8F E646F6378>
 MyDB6α のデータ更新のお願い 2012 年 4 月から薬価改正等が行われましたのでMyDBのバージョンアップとデータ更新をお願いします 始める前に自局の Xcodeを確認しておいてください Xcode は各薬局に特有の数字です 以前配布した MyDB の CD パッケージに記載されてありました わからないときは鹿児島県薬剤師会 (099)257-8288 にお問い合わせ下さい 新規で登録される方
MyDB6α のデータ更新のお願い 2012 年 4 月から薬価改正等が行われましたのでMyDBのバージョンアップとデータ更新をお願いします 始める前に自局の Xcodeを確認しておいてください Xcode は各薬局に特有の数字です 以前配布した MyDB の CD パッケージに記載されてありました わからないときは鹿児島県薬剤師会 (099)257-8288 にお問い合わせ下さい 新規で登録される方
内容 1 はじめに インストールの手順 起動の手順 Enterprise Architect のプロジェクトファイルを開く 内容を参照する プロジェクトブラウザを利用する ダイアグラムを開く 便利な機能.
 Viewer manual by SparxSystems Japan Enterprise Architect 読み込み専用版 (Viewer) 利用マニュアル 内容 1 はじめに...3 2 インストールの手順...3 3 起動の手順...6 4 Enterprise Architect のプロジェクトファイルを開く...7 5 内容を参照する...8 5.1 プロジェクトブラウザを利用する...8
Viewer manual by SparxSystems Japan Enterprise Architect 読み込み専用版 (Viewer) 利用マニュアル 内容 1 はじめに...3 2 インストールの手順...3 3 起動の手順...6 4 Enterprise Architect のプロジェクトファイルを開く...7 5 内容を参照する...8 5.1 プロジェクトブラウザを利用する...8
機能ゲノム学(第6回)
") トランスクリプトーム解析の今昔 なぜマイクロアレイ? なぜRNA-Seq? 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 Contents トランスクリプトーム解析の概要 各手法の長所 短所 マイクロアレイ
トランスクリプトーム解析の今昔 なぜマイクロアレイ? なぜRNA-Seq? 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 Contents トランスクリプトーム解析の概要 各手法の長所 短所 マイクロアレイ
目次 レジストリの設定...2 トレーディングソフトの自動起動設定...7 VPS 自動再起動の設定
 Windows サーバー 2008 R2 用 目次 レジストリの設定...2 トレーディングソフトの自動起動設定...7 VPS 自動再起動の設定...15 1 以下の作業工程の中に VPS またはトレーディングソフトの停止 再起動を行います お客様のお取引きに影響のない時間に作業を行ってください なお 以下の作業に伴う いかなる損失にも一切の責任を負いません また こちらのマニュアルは外部情報をもとに作成したものです
Windows サーバー 2008 R2 用 目次 レジストリの設定...2 トレーディングソフトの自動起動設定...7 VPS 自動再起動の設定...15 1 以下の作業工程の中に VPS またはトレーディングソフトの停止 再起動を行います お客様のお取引きに影響のない時間に作業を行ってください なお 以下の作業に伴う いかなる損失にも一切の責任を負いません また こちらのマニュアルは外部情報をもとに作成したものです
Microsoft Word - VB.doc
 第 1 章 初めてのプログラミング 本章では カウントアップというボタンを押すと表示されている値が1ずつ増加し カウントダウンというボタンを押すと表示されている値が1ずつ減少する簡単な機能のプログラムを作り これを通して Visual Basic.NET によるプログラム開発の概要を学んでいきます 1.1 起動とプロジェクトの新規作成 Visual Studio.NET の起動とプロジェクトの新規作成の方法を
第 1 章 初めてのプログラミング 本章では カウントアップというボタンを押すと表示されている値が1ずつ増加し カウントダウンというボタンを押すと表示されている値が1ずつ減少する簡単な機能のプログラムを作り これを通して Visual Basic.NET によるプログラム開発の概要を学んでいきます 1.1 起動とプロジェクトの新規作成 Visual Studio.NET の起動とプロジェクトの新規作成の方法を
PowerPoint プレゼンテーション
 FLEXSCHE Excel 帳票 入門ガイド 1 目次 2 EXCEL 帳票とは EDIF を用いて出力された一時データを元に それを EXCEL 形式の帳票として出力する機能です 利用するには FLEXSCHE EDIF の他 Microsoft Excel 2003 以降が必要です レイアウトデザインも EXCEL で行うので 多くの方に操作に抵抗なく編集していただけます この入門ガイドでは
FLEXSCHE Excel 帳票 入門ガイド 1 目次 2 EXCEL 帳票とは EDIF を用いて出力された一時データを元に それを EXCEL 形式の帳票として出力する機能です 利用するには FLEXSCHE EDIF の他 Microsoft Excel 2003 以降が必要です レイアウトデザインも EXCEL で行うので 多くの方に操作に抵抗なく編集していただけます この入門ガイドでは
Microsoft Word - macマニュアル【 】.doc
 目次 1. ログイン... 1 2. ログアウト... 3 3. デスクトップ ( 例 :Word Excel 起動中 )... 4 4. Dock( 例 :Word Excel 起動中 )... 5 5. Finder ウィンドウ... 9 6. メニューバー ( 例 :Word 起動中 )... 10 7. 文字の入力 ( 例 :Word で入力 )... 11 8. データの保存 ( 例 :Word
目次 1. ログイン... 1 2. ログアウト... 3 3. デスクトップ ( 例 :Word Excel 起動中 )... 4 4. Dock( 例 :Word Excel 起動中 )... 5 5. Finder ウィンドウ... 9 6. メニューバー ( 例 :Word 起動中 )... 10 7. 文字の入力 ( 例 :Word で入力 )... 11 8. データの保存 ( 例 :Word
コンピュータグラフィックス基礎 No
 課題 6: モデリング (1) OBJView の動作確認 ( レポートには含めなくてよい ) 次ページ以降の 課題用メモ を参考にして OBJ ファイルを 3D 表示する OBJView を実行し 画面に立体が表示されることを確認するとともに 以下の機能を確認しなさい 左ドラッグによる立体の回転 右ドラッグによる拡大/ 縮小 [v] キーによる頂点の表示 非表示 サンプルに含まれる bunny_3k.obj
課題 6: モデリング (1) OBJView の動作確認 ( レポートには含めなくてよい ) 次ページ以降の 課題用メモ を参考にして OBJ ファイルを 3D 表示する OBJView を実行し 画面に立体が表示されることを確認するとともに 以下の機能を確認しなさい 左ドラッグによる立体の回転 右ドラッグによる拡大/ 縮小 [v] キーによる頂点の表示 非表示 サンプルに含まれる bunny_3k.obj
PowerPoint プレゼンテーション
 バイオインフォマティクス 講習会 V 事前準備 が完了されている方は コンテナの起動 ファイルのコピー (Windows) まで 進めておいてください メニュー 1. 環境構築の確認 2. 基本的なLinuxコマンド 3. ツールのインストール 4. NGSデータの基礎知識と前処理 5. トランスクリプトのアッセンブル 6. RNA-seqのリファレンスcDNAマッピングとFPKM 算出 7. RNA-seqのリファレンスゲノムマッピングとFPKM
バイオインフォマティクス 講習会 V 事前準備 が完了されている方は コンテナの起動 ファイルのコピー (Windows) まで 進めておいてください メニュー 1. 環境構築の確認 2. 基本的なLinuxコマンド 3. ツールのインストール 4. NGSデータの基礎知識と前処理 5. トランスクリプトのアッセンブル 6. RNA-seqのリファレンスcDNAマッピングとFPKM 算出 7. RNA-seqのリファレンスゲノムマッピングとFPKM
SILAND.JP テンプレート集
 SILAND.JP のテンプレートを使った操作マニュアルの作成方法について スタイルの設定を使った文書作成 第 1 版 作成者しら 作成日 2014 年 2 月 21 日 最終更新日 2014 年 2 月 21 日 1 / 15 ダウンロードはこちら http://siland.jp/ 目次 SILAND.JP のテンプレートを使った操作マニュアルの作成方法について... 1 目次... 2 テンプレートのダウンロードについて...
SILAND.JP のテンプレートを使った操作マニュアルの作成方法について スタイルの設定を使った文書作成 第 1 版 作成者しら 作成日 2014 年 2 月 21 日 最終更新日 2014 年 2 月 21 日 1 / 15 ダウンロードはこちら http://siland.jp/ 目次 SILAND.JP のテンプレートを使った操作マニュアルの作成方法について... 1 目次... 2 テンプレートのダウンロードについて...
CASEC
 WEB 管理者マニュアル 2017 年 6 月版 お願い 本書で解説している 団体管理メニュー については 下記のパソコン環境のみ動作対応いたしております お手数ですが下記環境を満たしているパソコンにてご利用ください OS ブラウザ Windows 7 / 8.1 / 10 1 Internet Explorer 11, Google Chrome, Microsoft Edge20 以降 (Win
WEB 管理者マニュアル 2017 年 6 月版 お願い 本書で解説している 団体管理メニュー については 下記のパソコン環境のみ動作対応いたしております お手数ですが下記環境を満たしているパソコンにてご利用ください OS ブラウザ Windows 7 / 8.1 / 10 1 Internet Explorer 11, Google Chrome, Microsoft Edge20 以降 (Win
Microsoft PowerPoint - KanriManual.ppt
 環境一般教育 マニュアル 管理者用 1 管理者機能一覧管理者が利用できる機能は以下の通りです 印がついている操作手順について後のページでご説明いたします (p.10) 受講者の学習状況に応じメールを送信することができますメール設定 (p.16) 登録されている受講者の削除が出来ます受講者の削除受講者に関する操作 (p.5) 個人を指定して学習進捗を確認できます学習進捗 (p.3) 受講者を一覧にした受講状況を確認します
環境一般教育 マニュアル 管理者用 1 管理者機能一覧管理者が利用できる機能は以下の通りです 印がついている操作手順について後のページでご説明いたします (p.10) 受講者の学習状況に応じメールを送信することができますメール設定 (p.16) 登録されている受講者の削除が出来ます受講者の削除受講者に関する操作 (p.5) 個人を指定して学習進捗を確認できます学習進捗 (p.3) 受講者を一覧にした受講状況を確認します
RNA-seq
 RNA-seq 1 RNA-seq 解析フロー RNA-seq インポート クオリティチェック RNA-seq 発現差解析 この資料では RNA-seq からの説明となりますが インポート クオリティチェックについては サポート資料のページより内容をご確認いただけます 2 データ 発現解析用デモデータは 以下よりダウンロードいただけます ES 細胞 (ESC) と神経前駆細胞 (NPC) の発現解析を小さなデモデータで行えます
RNA-seq 1 RNA-seq 解析フロー RNA-seq インポート クオリティチェック RNA-seq 発現差解析 この資料では RNA-seq からの説明となりますが インポート クオリティチェックについては サポート資料のページより内容をご確認いただけます 2 データ 発現解析用デモデータは 以下よりダウンロードいただけます ES 細胞 (ESC) と神経前駆細胞 (NPC) の発現解析を小さなデモデータで行えます
C プログラミング演習 1( 再 ) 2 講義では C プログラミングの基本を学び 演習では やや実践的なプログラミングを通して学ぶ
 2 講義では C プログラミングの基本を学び 演習では やや実践的なプログラミングを通して学ぶ") C プログラミング演習 1( 再 ) 2 講義では C プログラミングの基本を学び 演習では やや実践的なプログラミングを通して学ぶ 今回のプログラミングの課題 次のステップによって 徐々に難易度の高いプログラムを作成する ( 参照用の番号は よくわかる C 言語 のページ番号 ) 1. キーボード入力された整数 10 個の中から最大のものを答える 2. 整数を要素とする配列 (p.57-59) に初期値を与えておき
C プログラミング演習 1( 再 ) 2 講義では C プログラミングの基本を学び 演習では やや実践的なプログラミングを通して学ぶ 今回のプログラミングの課題 次のステップによって 徐々に難易度の高いプログラムを作成する ( 参照用の番号は よくわかる C 言語 のページ番号 ) 1. キーボード入力された整数 10 個の中から最大のものを答える 2. 整数を要素とする配列 (p.57-59) に初期値を与えておき
目次 1. システム概要 設置手順 注意事項 動作環境 初期設定 システム設定 ( 環境設定 ) システム設定 ( ログインパスワード変更 ) システム設定 ( ファイルのパスワード変
 システム設定 ( ログインパスワード変更 ) システム設定 ( ファイルのパスワード変") 厚生労働省版ストレスチェック実施プログラム設置 設定マニュアル Ver.1.1 目次 1. システム概要... 2 2. 設置手順... 3 3. 注意事項... 5 4. 動作環境... 8 5. 初期設定... 9 6. システム設定 ( 環境設定 )... 15 7. システム設定 ( ログインパスワード変更 )... 18 8. システム設定 ( ファイルのパスワード変更 )... 20 9.
厚生労働省版ストレスチェック実施プログラム設置 設定マニュアル Ver.1.1 目次 1. システム概要... 2 2. 設置手順... 3 3. 注意事項... 5 4. 動作環境... 8 5. 初期設定... 9 6. システム設定 ( 環境設定 )... 15 7. システム設定 ( ログインパスワード変更 )... 18 8. システム設定 ( ファイルのパスワード変更 )... 20 9.
Outlook2010 の メール 連絡先 に関連する内容を解説します 注意 :Outlook2007 と Outlook2010 では 基本操作 基本画面が違うため この資料では Outlook2010 のみで参考にしてください Outlook2010 の画面構成について... 2 メールについて
 Outlook2010 - メール 連絡先など - Outlook2010 の メール 連絡先 に関連する内容を解説します 注意 :Outlook2007 と Outlook2010 では 基本操作 基本画面が違うため この資料では Outlook2010 のみで参考にしてください Outlook2010 の画面構成について... 2 メールについて... 3 画面構成と操作... 3 人物情報ウィンドウ...
Outlook2010 - メール 連絡先など - Outlook2010 の メール 連絡先 に関連する内容を解説します 注意 :Outlook2007 と Outlook2010 では 基本操作 基本画面が違うため この資料では Outlook2010 のみで参考にしてください Outlook2010 の画面構成について... 2 メールについて... 3 画面構成と操作... 3 人物情報ウィンドウ...
2. オプション設定画面で, 必要事項を記入 選択します. 少なくとも, タイトル に課題の見出しとなる文章を入力する他, 種別 を アンケート( 無記名式 ) に設定する必要があります. また, アクセス制限はここでは コースメニューで非表示にする に設定します. その他設定は必要に応じて行って下
 に設定する必要があります. また, アクセス制限はここでは コースメニューで非表示にする に設定します. その他設定は必要に応じて行って下") (WebClass チュートリアル ) 公開アンケートの実施 ここではアンケート, 特にメンバーを限定せず広く実施する無記名アンケート ( 以下, 公開アンケート ) の実施方法について解説します. 公開アンケートでは, 回答者が WebClass にログインすることなく回答できるというメリットがありますが, 回答資格の判別や, 同一人による複数回の回答をチェックすることが出来ない欠点がありますのでご注意下さい.
(WebClass チュートリアル ) 公開アンケートの実施 ここではアンケート, 特にメンバーを限定せず広く実施する無記名アンケート ( 以下, 公開アンケート ) の実施方法について解説します. 公開アンケートでは, 回答者が WebClass にログインすることなく回答できるというメリットがありますが, 回答資格の判別や, 同一人による複数回の回答をチェックすることが出来ない欠点がありますのでご注意下さい.
このうち ツールバーが表示されていないときは メニューバーから [ 表示 (V)] [ ツールバー (T)] の [ 標準のボタン (S)] [ アドレスバー (A)] と [ ツールバーを固定する (B)] をクリックしてチェックを付けておくとよい また ツールバーはユーザ ( 利用者 ) が変更
![このうち ツールバーが表示されていないときは メニューバーから [ 表示 (V)] [ ツールバー (T)] の [ 標準のボタン (S)] [ アドレスバー (A)] と [ ツールバーを固定する (B)] をクリックしてチェックを付けておくとよい また ツールバーはユーザ ( 利用者 ) が変更](/thumbs/93/112077293.jpg "このうち ツールバーが表示されていないときは メニューバーから [ 表示 (V)] [ ツールバー (T)] の [ 標準のボタン (S)] [ アドレスバー (A)] と [ ツールバーを固定する (B)] をクリックしてチェックを付けておくとよい また ツールバーはユーザ ( 利用者 ) が変更") ファイル操作 アプリケーションソフトウェアなどで作成したデータはディスクにファイルとして保存される そのファイルに関してコピーや削除などの基本的な操作について実習する また ファイルを整理するためのフォルダの作成などの実習をする (A) ファイル名 ファイル名はデータなどのファイルをディスクに保存しておくときに付ける名前である データファイルはどんどん増えていくので 何のデータであるのかわかりやすいファイル名を付けるようにする
ファイル操作 アプリケーションソフトウェアなどで作成したデータはディスクにファイルとして保存される そのファイルに関してコピーや削除などの基本的な操作について実習する また ファイルを整理するためのフォルダの作成などの実習をする (A) ファイル名 ファイル名はデータなどのファイルをディスクに保存しておくときに付ける名前である データファイルはどんどん増えていくので 何のデータであるのかわかりやすいファイル名を付けるようにする
分析のステップ Step 1: Y( 目的変数 ) に対する値の順序を確認 Step 2: モデルのあてはめ を実行 適切なモデルの指定 Step 3: オプションを指定し オッズ比とその信頼区間を表示 以下 このステップに沿って JMP の操作をご説明します Step 1: Y( 目的変数 ) の
 に対する値の順序を確認 Step 2: モデルのあてはめ を実行 適切なモデルの指定 Step 3: オプションを指定し オッズ比とその信頼区間を表示 以下 このステップに沿って JMP の操作をご説明します Step 1: Y( 目的変数 ) の") JMP によるオッズ比 リスク比 ( ハザード比 ) の算出と注意点 SAS Institute Japan 株式会社 JMP ジャパン事業部 2011 年 10 月改定 1. はじめに 本文書は JMP でロジスティック回帰モデルによるオッズ比 比例ハザードモデルによるリスク比 それぞれに対する信頼区間を求める操作方法と注意点を述べたものです 本文書は JMP 7 以降のバージョンに対応しております
JMP によるオッズ比 リスク比 ( ハザード比 ) の算出と注意点 SAS Institute Japan 株式会社 JMP ジャパン事業部 2011 年 10 月改定 1. はじめに 本文書は JMP でロジスティック回帰モデルによるオッズ比 比例ハザードモデルによるリスク比 それぞれに対する信頼区間を求める操作方法と注意点を述べたものです 本文書は JMP 7 以降のバージョンに対応しております
Maser - User Operation Manual
 Maser 3 Cell Innovation User Operation Manual 2013.4.1 1 目次 1. はじめに... 3 1.1. 推奨動作環境... 3 2. データの登録... 4 2.1. プロジェクトの作成... 4 2.2. Projectへのデータのアップロード... 8 2.2.1. HTTPSでのアップロード... 8 2.2.2. SFTPでのアップロード...
Maser 3 Cell Innovation User Operation Manual 2013.4.1 1 目次 1. はじめに... 3 1.1. 推奨動作環境... 3 2. データの登録... 4 2.1. プロジェクトの作成... 4 2.2. Projectへのデータのアップロード... 8 2.2.1. HTTPSでのアップロード... 8 2.2.2. SFTPでのアップロード...
Shareresearchオンラインマニュアル
 Chrome の初期設定 以下の手順で設定してください 1. ポップアップブロックの設定 2. 推奨する文字サイズの設定 3. 規定のブラウザに設定 4. ダウンロードファイルの保存先の設定 5.PDFレイアウトの印刷設定 6. ランキングやハイライトの印刷設定 7. 注意事項 なお 本マニュアルの内容は バージョン 61.0.3163.79 の Chrome を基に説明しています Chrome の設定手順や画面については
Chrome の初期設定 以下の手順で設定してください 1. ポップアップブロックの設定 2. 推奨する文字サイズの設定 3. 規定のブラウザに設定 4. ダウンロードファイルの保存先の設定 5.PDFレイアウトの印刷設定 6. ランキングやハイライトの印刷設定 7. 注意事項 なお 本マニュアルの内容は バージョン 61.0.3163.79 の Chrome を基に説明しています Chrome の設定手順や画面については
目 次 1. はじめに ソフトの起動と終了 環境設定 発助 SMS ファイルの操作 電話番号設定 運用条件 回線情報 SMS 送信の開始と停止 ファイル出力... 16
 発助 SMS 操作マニュアル Ver1.2 2018.7.21 ソフトプラン合同会社 1/18 目 次 1. はじめに... 3 2. ソフトの起動と終了... 3 3. 環境設定... 5 4. 発助 SMS ファイルの操作... 7 5. 電話番号設定... 9 6. 運用条件... 11 7. 回線情報... 12 8.SMS 送信の開始と停止... 13 9. ファイル出力... 16 10.
発助 SMS 操作マニュアル Ver1.2 2018.7.21 ソフトプラン合同会社 1/18 目 次 1. はじめに... 3 2. ソフトの起動と終了... 3 3. 環境設定... 5 4. 発助 SMS ファイルの操作... 7 5. 電話番号設定... 9 6. 運用条件... 11 7. 回線情報... 12 8.SMS 送信の開始と停止... 13 9. ファイル出力... 16 10.
CLC Genomics Workbench ウェブトレーニングセミナー: 変異解析編
 CLC Genomics Workbench ウェブトレーニングセミナー : 遺伝子発現解析編 12 th Feb., 2016 フィルジェン株式会社バイオサイエンス部 [email protected] Feb., 2016_V2 1 遺伝子発現解析概要 本日のセミナーにおける解析の流れ及び使用するツール名 ( 図中赤枠部分 ) Case Control インポート インポート インポート
CLC Genomics Workbench ウェブトレーニングセミナー : 遺伝子発現解析編 12 th Feb., 2016 フィルジェン株式会社バイオサイエンス部 [email protected] Feb., 2016_V2 1 遺伝子発現解析概要 本日のセミナーにおける解析の流れ及び使用するツール名 ( 図中赤枠部分 ) Case Control インポート インポート インポート
IonTorrent RNA-Seq 解析概要 サーモフィッシャーサイエンティフィックライフテクノロジーズジャパンテクニカルサポート The world leader in serving science
 IonTorrent RNA-Seq 解析概要 2017-03 サーモフィッシャーサイエンティフィックライフテクノロジーズジャパンテクニカルサポート The world leader in serving science 資料概要 この資料は IonTorrent シーケンサーで RNA-Seq (WholeTranscriptome mrna ampliseqrna mirna) 解析を実施されるユーザー様向けの内容となっています
IonTorrent RNA-Seq 解析概要 2017-03 サーモフィッシャーサイエンティフィックライフテクノロジーズジャパンテクニカルサポート The world leader in serving science 資料概要 この資料は IonTorrent シーケンサーで RNA-Seq (WholeTranscriptome mrna ampliseqrna mirna) 解析を実施されるユーザー様向けの内容となっています
目次 1. HLA Fusion 3.0 がインストール可能な環境 HLA Fusion 3.0 のインストール HLA Fusion 3.4 のインストール 初期設定用データベース接続 ( 初めての方のみ ) 既存データベースのUpg
 既存データベースのUpg") 目次 1. HLA Fusion 3.0 がインストール可能な環境... 1 2. HLA Fusion 3.0 のインストール... 2 3. HLA Fusion 3.4 のインストール... 4 4. 初期設定用データベース接続 ( 初めての方のみ )... 5 5. 既存データベースのUpgrade 方法 (HLA Fusion 3~3.3 を既に使用の方 )... 7 6. インストールが成功したかの確認...
目次 1. HLA Fusion 3.0 がインストール可能な環境... 1 2. HLA Fusion 3.0 のインストール... 2 3. HLA Fusion 3.4 のインストール... 4 4. 初期設定用データベース接続 ( 初めての方のみ )... 5 5. 既存データベースのUpgrade 方法 (HLA Fusion 3~3.3 を既に使用の方 )... 7 6. インストールが成功したかの確認...
スクールCOBOL2002
 3. 関連資料 - よく使われる機能の操作方法 - (a) ファイルの入出力処理 - 順ファイル等を使ったプログラムの実行 - - 目次 -. はじめに 2. コーディング上の指定 3. 順ファイルの使用方法 4. プリンタへの出力方法 5. 索引ファイルの使用方法 6. 終わりに 2 . はじめに 本説明書では 簡単なプログラム ( ファイル等を使わないプログラム ) の作成からコンパイル 実行までの使用方法は既に理解しているものとして
3. 関連資料 - よく使われる機能の操作方法 - (a) ファイルの入出力処理 - 順ファイル等を使ったプログラムの実行 - - 目次 -. はじめに 2. コーディング上の指定 3. 順ファイルの使用方法 4. プリンタへの出力方法 5. 索引ファイルの使用方法 6. 終わりに 2 . はじめに 本説明書では 簡単なプログラム ( ファイル等を使わないプログラム ) の作成からコンパイル 実行までの使用方法は既に理解しているものとして
目次 1. Azure Storage をインストールする Azure Storage のインストール Azure Storage のアンインストール Azure Storage を使う ストレージアカウントの登録... 7
 QNAP Azure Storage ユーザーガイド 発行 : 株式会社フォースメディア 2014/6/2 Rev. 1.00 2014 Force Media, Inc. 目次 1. Azure Storage をインストールする... 3 1.1. Azure Storage のインストール... 3 1.2. Azure Storage のアンインストール... 5 2. Azure Storage
QNAP Azure Storage ユーザーガイド 発行 : 株式会社フォースメディア 2014/6/2 Rev. 1.00 2014 Force Media, Inc. 目次 1. Azure Storage をインストールする... 3 1.1. Azure Storage のインストール... 3 1.2. Azure Storage のアンインストール... 5 2. Azure Storage
PowerPoint プレゼンテーション
 JAN コード登録マニュアル 項目説明 CSV で商品データを upload するに当たり 間違えやすいカラムについてまとめました 項目 説明 備考 コントロールカラム CSV 上で当該商品情報をどうするのか ( 更新 削除等 ) 指示するコード "u": 更新 "d": 削除等 商品管理番号 出来上がった商品ページURLの一部であり 入力がない場合は自動採番される web 上で商品を特定するキーコード
JAN コード登録マニュアル 項目説明 CSV で商品データを upload するに当たり 間違えやすいカラムについてまとめました 項目 説明 備考 コントロールカラム CSV 上で当該商品情報をどうするのか ( 更新 削除等 ) 指示するコード "u": 更新 "d": 削除等 商品管理番号 出来上がった商品ページURLの一部であり 入力がない場合は自動採番される web 上で商品を特定するキーコード
講義内容 ファイル形式 データの可視化 データのクオリティチェック マッピング アセンブル 資料の見方 $ pwd 実際に入力するコマンドを黄色い四角の中に示します 2
 N G S 解析基礎 講義内容 ファイル形式 データの可視化 データのクオリティチェック マッピング アセンブル 資料の見方 $ pwd 実際に入力するコマンドを黄色い四角の中に示します 2 ファイル形式 NGS 解析でよく使われるファイル形式 ファイル形式 fastq bam/sam vcf bed fasta サンプルデータの場所 /home/ ユーザ名 /Desktop/amelieff/1K_ERR038793_1.fastq
N G S 解析基礎 講義内容 ファイル形式 データの可視化 データのクオリティチェック マッピング アセンブル 資料の見方 $ pwd 実際に入力するコマンドを黄色い四角の中に示します 2 ファイル形式 NGS 解析でよく使われるファイル形式 ファイル形式 fastq bam/sam vcf bed fasta サンプルデータの場所 /home/ ユーザ名 /Desktop/amelieff/1K_ERR038793_1.fastq
PowerPoint プレゼンテーション
 V1 次世代シークエンサ実習 II 本講義にあたって 代表的な解析の流れを紹介します 論文でよく使用されているツールを使用します コマンドを沢山実行します スペルミスが心配な方は コマンド例がありますのでコピーして実行してください /home/admin1409/amelieff/ngs/reseq_command.txt マークのコマンドは実行してください 実行が遅れてもあせらずに 応用や課題の間に追い付いてください
V1 次世代シークエンサ実習 II 本講義にあたって 代表的な解析の流れを紹介します 論文でよく使用されているツールを使用します コマンドを沢山実行します スペルミスが心配な方は コマンド例がありますのでコピーして実行してください /home/admin1409/amelieff/ngs/reseq_command.txt マークのコマンドは実行してください 実行が遅れてもあせらずに 応用や課題の間に追い付いてください
ガイダンス
 情報科学 B 第 2 回変数 1 今日やること Java プログラムの書き方 変数とは何か? 2 Java プログラムの書き方 3 作業手順 Java 言語を用いてソースコードを記述する (Cpad エディタを使用 ) コンパイル (Cpad エディタを使用 ) 実行 (Cpad エディタを使用 ) エラーが出たらどうしたらよいか??? 4 書き方 これから作成する Hello.java 命令文 メソッドブロック
情報科学 B 第 2 回変数 1 今日やること Java プログラムの書き方 変数とは何か? 2 Java プログラムの書き方 3 作業手順 Java 言語を用いてソースコードを記述する (Cpad エディタを使用 ) コンパイル (Cpad エディタを使用 ) 実行 (Cpad エディタを使用 ) エラーが出たらどうしたらよいか??? 4 書き方 これから作成する Hello.java 命令文 メソッドブロック
NGS速習コース
 バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-4. R Bioconductor I 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) [email protected] http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-4. R Bioconductor I 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) [email protected] http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
カルテダウンロード 操作マニュアル
 カルテ ZERO 操作マニュアル カルテダウンロード Ver1. 3 目 次 カルテダウンロード カルテダウンロード時の注意点 1. インストール 2. カルテダウンロード 2-1. 時間を設定し自動でダウンロードする方法 2-2. 手動でダウンロードする方法 3. 補足説明 P.3 P.4 P.9 P.14 P.18 P.20 カルテダウンロード時の注意点 カルテダウンロードは Windows 7
カルテ ZERO 操作マニュアル カルテダウンロード Ver1. 3 目 次 カルテダウンロード カルテダウンロード時の注意点 1. インストール 2. カルテダウンロード 2-1. 時間を設定し自動でダウンロードする方法 2-2. 手動でダウンロードする方法 3. 補足説明 P.3 P.4 P.9 P.14 P.18 P.20 カルテダウンロード時の注意点 カルテダウンロードは Windows 7
内容 MD00Manager とは?... MD00Manager をインストールする.... ソフトのインストール... MD00Manager の使い方.... 起動をする... 機能説明...7 機能説明 ( メニューバー )...8 機能説明 ( ステータスバー )...8 機能説明 ( コ
...8 機能説明 ( ステータスバー )...8 機能説明 ( コ") MD00Manager マニュアル このマニュアルでは MD00Manager の詳細設定を行うことが出来ます MD00Manager では MD00 の設定変更や読み取ったデータを CSV に出力 照合用のデータベースの作成を行えます MD00 についてはそれぞれのマニュアルを確認して下さい Ver.0 内容 MD00Manager とは?... MD00Manager をインストールする....
MD00Manager マニュアル このマニュアルでは MD00Manager の詳細設定を行うことが出来ます MD00Manager では MD00 の設定変更や読み取ったデータを CSV に出力 照合用のデータベースの作成を行えます MD00 についてはそれぞれのマニュアルを確認して下さい Ver.0 内容 MD00Manager とは?... MD00Manager をインストールする....
スライド 1
 適用マニュアル Hos-CanR 3.0 サービスパック適用マニュアル システム管理者用 SP1.4 バージョン 改訂日付 改訂内容 SP 1.4 2011/12/05 SP1.4リリースに伴う修正 SP 1.3 2011/11/01 リリースに伴う修正 SP 1.2 2010/12/10 SP1.2リリースに伴う修正 SP 1.1 2010/08/09 SP1.1リリースに伴う修正 SP 1.0
適用マニュアル Hos-CanR 3.0 サービスパック適用マニュアル システム管理者用 SP1.4 バージョン 改訂日付 改訂内容 SP 1.4 2011/12/05 SP1.4リリースに伴う修正 SP 1.3 2011/11/01 リリースに伴う修正 SP 1.2 2010/12/10 SP1.2リリースに伴う修正 SP 1.1 2010/08/09 SP1.1リリースに伴う修正 SP 1.0
目次 第 1 章はじめに 本ソフトの概要... 2 第 2 章インストール編 ソフトの動作環境を確認しましょう ソフトをコンピュータにセットアップしましょう 動作を確認しましょう コンピュータからアンインストー
 JS 管理ファイル作成支援ソフト 工事用 Ver.4.0 インストールマニュアル 操作マニュアル 日本下水道事業団 目次 第 1 章はじめに... 1 1-1 本ソフトの概要... 2 第 2 章インストール編... 3 2-1 ソフトの動作環境を確認しましょう... 4 2-2 ソフトをコンピュータにセットアップしましょう... 5 2-3 動作を確認しましょう... 8 2-4 コンピュータからアンインストールする方法...
JS 管理ファイル作成支援ソフト 工事用 Ver.4.0 インストールマニュアル 操作マニュアル 日本下水道事業団 目次 第 1 章はじめに... 1 1-1 本ソフトの概要... 2 第 2 章インストール編... 3 2-1 ソフトの動作環境を確認しましょう... 4 2-2 ソフトをコンピュータにセットアップしましょう... 5 2-3 動作を確認しましょう... 8 2-4 コンピュータからアンインストールする方法...
dae opixrae 1 Feb Mar Apr May Jun と表示される 今 必要なのは opixrae のデータだけなので > opixrae=opixdaa$opi
 R による時系列分析 4 1. GARCH モデルを推定する 1.1 パッケージ rugarch をインスツールする パッケージとは通常の R には含まれていない 追加的な R のコマンドの集まりのようなものである R には追加的に 600 以上のパッケージが用意されており それぞれ分析の目的に応じて標準の R にパッケージを追加していくことになる インターネットに接続してあるパソコンで R を起動させ
R による時系列分析 4 1. GARCH モデルを推定する 1.1 パッケージ rugarch をインスツールする パッケージとは通常の R には含まれていない 追加的な R のコマンドの集まりのようなものである R には追加的に 600 以上のパッケージが用意されており それぞれ分析の目的に応じて標準の R にパッケージを追加していくことになる インターネットに接続してあるパソコンで R を起動させ
Fortinet 社 FortiExplorer 操作マニュアル 株式会社ネットワークバリューコンポネンツ 第一版 Page1 Network Value Components Ltd. Copyright (c)2012 Network Value Components Ltd. All Righ
2012 Network Value Components Ltd. All Righ") Fortinet 社 FortiExplorer 操作マニュアル 株式会社ネットワークバリューコンポネンツ 第一版 Page1 Copyright (c)2012 All Right Reserved. 目次 1 ソフトウェアダウンロード... 3 1.1 PC(Mac, Windows)... 3 2 ソフトウェアインストール... 4 2.1 Mac... 4 2.2 Windows... 6
Fortinet 社 FortiExplorer 操作マニュアル 株式会社ネットワークバリューコンポネンツ 第一版 Page1 Copyright (c)2012 All Right Reserved. 目次 1 ソフトウェアダウンロード... 3 1.1 PC(Mac, Windows)... 3 2 ソフトウェアインストール... 4 2.1 Mac... 4 2.2 Windows... 6
電子納品チェックシステム利用マニュアル
 香川県版 Calcheck チェック専用版 利用マニュアル 目 次 1. 概要... 1 1-1 システムの基本機能... 1 1-2 システムの機能概要... 1 1-2-1 対応する要領 基準... 1 1-2-2 動作環境... 1 1-2-3 電子納品データのチェック手順... 2 2. インストール... 3 2-1 システムのインストール... 3 2-2 バージョンアップ... 6 2-2-1
香川県版 Calcheck チェック専用版 利用マニュアル 目 次 1. 概要... 1 1-1 システムの基本機能... 1 1-2 システムの機能概要... 1 1-2-1 対応する要領 基準... 1 1-2-2 動作環境... 1 1-2-3 電子納品データのチェック手順... 2 2. インストール... 3 2-1 システムのインストール... 3 2-2 バージョンアップ... 6 2-2-1
PowerPoint プレゼンテーション
 かんたんマニュアル 差し込み印刷編 目次 STEP:1 STEP:2 STEP:3 STEP:4 元となるラベル カードのデータを作ろうP.2 差し込みデータの関連付けを設定しよう P.7 データの差し込みをしよう P.11 印刷しよう P.17 STEP1: 画面の確認をしよう 差し込み印刷とは 表計算ソフトで作った住所録を宛名ラベルに印刷したり 名簿をも とに同じ形式のカードを作ったりするときに便利な機能です
かんたんマニュアル 差し込み印刷編 目次 STEP:1 STEP:2 STEP:3 STEP:4 元となるラベル カードのデータを作ろうP.2 差し込みデータの関連付けを設定しよう P.7 データの差し込みをしよう P.11 印刷しよう P.17 STEP1: 画面の確認をしよう 差し込み印刷とは 表計算ソフトで作った住所録を宛名ラベルに印刷したり 名簿をも とに同じ形式のカードを作ったりするときに便利な機能です
GWB
 NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 参照ゲノム配列へのマッピング 再アライメント 変異検出 CLC Genomics Workbench または Biomedical Genomics Workbench
NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 参照ゲノム配列へのマッピング 再アライメント 変異検出 CLC Genomics Workbench または Biomedical Genomics Workbench
プログラミング基礎
 C プログラミング Ⅰ 授業ガイダンス C 言語の概要プログラム作成 実行方法 授業内容について 授業目的 C 言語によるプログラミングの基礎を学ぶこと 学習内容 C 言語の基礎的な文法 入出力, 変数, 演算, 条件分岐, 繰り返し, 配列,( 関数 ) C 言語による簡単な計算処理プログラムの開発 到達目標 C 言語の基礎的な文法を理解する 簡単な計算処理プログラムを作成できるようにする 授業ガイダンス
C プログラミング Ⅰ 授業ガイダンス C 言語の概要プログラム作成 実行方法 授業内容について 授業目的 C 言語によるプログラミングの基礎を学ぶこと 学習内容 C 言語の基礎的な文法 入出力, 変数, 演算, 条件分岐, 繰り返し, 配列,( 関数 ) C 言語による簡単な計算処理プログラムの開発 到達目標 C 言語の基礎的な文法を理解する 簡単な計算処理プログラムを作成できるようにする 授業ガイダンス
レポート作成に役立つWord2013の機能
 レポート作成に役立つ Word2013 の機能 神戸親和女子大学情報処理教育センター 2015 年 10 月版 目次 1 文字数や行数を指定する... 2 2 ページ番号... 3 2.1 ページ番号をつける... 3 2.2 先頭ページだけページ番号を非表示にする... 4 2.3 3 ページ目からページ番号をつける... 5 2.4 ページ番号の開始番号を指定する... 7 3 目次の作成 更新...
レポート作成に役立つ Word2013 の機能 神戸親和女子大学情報処理教育センター 2015 年 10 月版 目次 1 文字数や行数を指定する... 2 2 ページ番号... 3 2.1 ページ番号をつける... 3 2.2 先頭ページだけページ番号を非表示にする... 4 2.3 3 ページ目からページ番号をつける... 5 2.4 ページ番号の開始番号を指定する... 7 3 目次の作成 更新...
目次 1. ログイン ログアウト デスクトップ ( 例 :Word Excel 起動中 ) Dock( 例 :Word Excel 起動中 ) Finder ウィンドウ メニューバー ( 例 :Word 起動中 )...
 Dock( 例 :Word Excel 起動中 ) Finder ウィンドウ メニューバー ( 例 :Word 起動中 )...") 2017 年 9 月 19 日 目次 1. ログイン... 1 2. ログアウト... 3 3. デスクトップ ( 例 :Word Excel 起動中 )... 4 4. Dock( 例 :Word Excel 起動中 )... 5 5. Finder ウィンドウ... 9 6. メニューバー ( 例 :Word 起動中 )... 10 7. 文字の入力 ( 例 :Word で入力 )... 11
2017 年 9 月 19 日 目次 1. ログイン... 1 2. ログアウト... 3 3. デスクトップ ( 例 :Word Excel 起動中 )... 4 4. Dock( 例 :Word Excel 起動中 )... 5 5. Finder ウィンドウ... 9 6. メニューバー ( 例 :Word 起動中 )... 10 7. 文字の入力 ( 例 :Word で入力 )... 11
エンドポイント濁度測定装置 LT-16 取扱説明書
 エンドポイント濁度測定装置 LT-16 LT-16 Manager マニュアル ( 簡易マニュアル Version 2.0) LT-16 Manager のインストール LT-16 Manager は添付の CD に内蔵されています LT-16 Manager は Windows 7 Windows 8 において動作確認をしております ( 以下の図は Windows 8 使用時の表示図面です ) ただし
エンドポイント濁度測定装置 LT-16 LT-16 Manager マニュアル ( 簡易マニュアル Version 2.0) LT-16 Manager のインストール LT-16 Manager は添付の CD に内蔵されています LT-16 Manager は Windows 7 Windows 8 において動作確認をしております ( 以下の図は Windows 8 使用時の表示図面です ) ただし
<4D F736F F F696E74202D20352D335F8D5C90AC CF909482CC90B690AC82C695D28F572E707074>
 RD_301 構成要素一覧と検索 から構成要素の編集辞書 ( 削除 ) を作る 作成 ( 編集 ) する削除辞書を開きます 構成要素を検索します ドラック & ドロップでも OK 範囲を選択して右クリック 右クリック 削除辞書に登録 ( 追加 ) したい構成要素を選択しコピーします 削除辞書に追加 ( 貼りつけ ) ます Step5. 削除辞書に構成要素が登録 ( 追加 ) されます 構成要素一覧と検索
RD_301 構成要素一覧と検索 から構成要素の編集辞書 ( 削除 ) を作る 作成 ( 編集 ) する削除辞書を開きます 構成要素を検索します ドラック & ドロップでも OK 範囲を選択して右クリック 右クリック 削除辞書に登録 ( 追加 ) したい構成要素を選択しコピーします 削除辞書に追加 ( 貼りつけ ) ます Step5. 削除辞書に構成要素が登録 ( 追加 ) されます 構成要素一覧と検索
電子納品チェックシステム利用マニュアル
 高知県版電子納品チェックシステム 利用マニュアル Ver.15 高知県 目 次 1. 概要... 1 1-1 システムの基本機能... 1 1-2 システムの機能概要... 1 1-2-1 対応する要領 基準... 1 1-2-2 動作環境... 1 1-2-3 電子納品データのチェック手順... 2 2. インストール... 3 2-1 システムのインストール... 3 2-2 バージョンアップ...
高知県版電子納品チェックシステム 利用マニュアル Ver.15 高知県 目 次 1. 概要... 1 1-1 システムの基本機能... 1 1-2 システムの機能概要... 1 1-2-1 対応する要領 基準... 1 1-2-2 動作環境... 1 1-2-3 電子納品データのチェック手順... 2 2. インストール... 3 2-1 システムのインストール... 3 2-2 バージョンアップ...
目次 第 1 章はじめに 本ソフトの概要... 2 第 2 章インストール編 ソフトの動作環境を確認しましょう ソフトをコンピュータにセットアップしましょう 動作を確認しましょう コンピュータからアンインストー
 JS 管理ファイル作成支援ソフト 設計用 Ver.4.0 インストールマニュアル 操作マニュアル 日本下水道事業団 目次 第 1 章はじめに... 1 1-1 本ソフトの概要... 2 第 2 章インストール編... 3 2-1 ソフトの動作環境を確認しましょう... 4 2-2 ソフトをコンピュータにセットアップしましょう... 5 2-3 動作を確認しましょう... 9 2-4 コンピュータからアンインストールする方法...
JS 管理ファイル作成支援ソフト 設計用 Ver.4.0 インストールマニュアル 操作マニュアル 日本下水道事業団 目次 第 1 章はじめに... 1 1-1 本ソフトの概要... 2 第 2 章インストール編... 3 2-1 ソフトの動作環境を確認しましょう... 4 2-2 ソフトをコンピュータにセットアップしましょう... 5 2-3 動作を確認しましょう... 9 2-4 コンピュータからアンインストールする方法...
Rでゲノム・トランスクリプトーム解析
 06.08. 版 スライド 8 までは自習 当日はスライド 9 から始める予定 スライド 3-86 は当日省略予定 講習会後に各自で復習してください 第 3 部 :NGS 解析 ( 中 ~ 上級 ) ~ トランスクリプトームアセンブリ 発現量推定 ~ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected]
06.08. 版 スライド 8 までは自習 当日はスライド 9 から始める予定 スライド 3-86 は当日省略予定 講習会後に各自で復習してください 第 3 部 :NGS 解析 ( 中 ~ 上級 ) ~ トランスクリプトームアセンブリ 発現量推定 ~ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected]
IPPO - 校内研修支援プログラム - 使用説明書 目次 項 目 ページ 1 プログラム利用の準備 この説明書の記述について プログラムの動作環境等 プログラムファイルのコピー プログラムファイルの起動 4 2 プログラムファイルの利用
 IPPO - 校内研修支援プログラム - 使用説明書 目次 項 目 ページ 1 プログラム利用の準備 1 1-1 この説明書の記述について 1 1-2 プログラムの動作環境等 1 1-3 プログラムファイルのコピー 1 1-4 プログラムファイルの起動 4 2 プログラムファイルの利用 5 2-1 スタート画面 5 2-2 各ボタンの説明 ( 機能概要 ) 6 3 児童 ( 生徒 ) 出席番号 氏名管理の入力
IPPO - 校内研修支援プログラム - 使用説明書 目次 項 目 ページ 1 プログラム利用の準備 1 1-1 この説明書の記述について 1 1-2 プログラムの動作環境等 1 1-3 プログラムファイルのコピー 1 1-4 プログラムファイルの起動 4 2 プログラムファイルの利用 5 2-1 スタート画面 5 2-2 各ボタンの説明 ( 機能概要 ) 6 3 児童 ( 生徒 ) 出席番号 氏名管理の入力
Microsoft Word - CygwinでPython.docx
 Cygwin でプログラミング 2018/4/9 千葉 数値計算は計算プログラムを書いて行うわけですが プログラムには様々な 言語 があるので そのうちどれかを選択する必要があります プログラム言語には 人間が書いたプログラムを一度計算機用に翻訳したのち計算を実行するものと 人間が書いたプログラムを計算機が読んでそのまま実行するものとがあります ( 若干不正確な説明ですが ) 前者を システム言語
Cygwin でプログラミング 2018/4/9 千葉 数値計算は計算プログラムを書いて行うわけですが プログラムには様々な 言語 があるので そのうちどれかを選択する必要があります プログラム言語には 人間が書いたプログラムを一度計算機用に翻訳したのち計算を実行するものと 人間が書いたプログラムを計算機が読んでそのまま実行するものとがあります ( 若干不正確な説明ですが ) 前者を システム言語
厚生労働省版ストレスチェック実施プログラム 設置 設定マニュアル Ver.3.2 目次 1. プログラム概要 注意事項 動作環境 設置 設定の流れ 設置手順 要注意 zip ファイル解凍の準備 実施者用管
 厚生労働省版ストレスチェック実施プログラム 設置 設定マニュアル Ver.3.2 目次 1. プログラム概要... 2 2. 注意事項... 3 3. 動作環境... 4 4. 設置 設定の流れ... 5 5. 設置手順... 6 6. 要注意 zip ファイル解凍の準備... 7 7. 実施者用管理ツール初期設定... 8 8. Windows によって PC が保護されました というダイアログが表示される場合...
厚生労働省版ストレスチェック実施プログラム 設置 設定マニュアル Ver.3.2 目次 1. プログラム概要... 2 2. 注意事項... 3 3. 動作環境... 4 4. 設置 設定の流れ... 5 5. 設置手順... 6 6. 要注意 zip ファイル解凍の準備... 7 7. 実施者用管理ツール初期設定... 8 8. Windows によって PC が保護されました というダイアログが表示される場合...
UNITAMA採点登録ガイド xlsx
 UNITAMA 採点登録ガイド 教学部授業運営課 2017 年 6 月更新 1-1. 採点授業一覧 から 評価をする授業を選択しクリックしてください 評価をつけたい授業を選択します 1-2. 採点登録 画面が開きますので 評価欄の箇所にチェックを入れていきます 3 2 4 1 5 6 画面説明 1 評価 素点 ( 単位互換のみ ) 出席率 理由を入れてください < 評価 > 本学では評価 (S~F)
UNITAMA 採点登録ガイド 教学部授業運営課 2017 年 6 月更新 1-1. 採点授業一覧 から 評価をする授業を選択しクリックしてください 評価をつけたい授業を選択します 1-2. 採点登録 画面が開きますので 評価欄の箇所にチェックを入れていきます 3 2 4 1 5 6 画面説明 1 評価 素点 ( 単位互換のみ ) 出席率 理由を入れてください < 評価 > 本学では評価 (S~F)
Microsoft Word - 動画が視聴できない場合.docx
 動画が視聴できない場合 動作 推奨環境を満たし必要なソフトウェアもインストールしたうえで プレビュー動 画が再生できない場合は以下を確認してください 動作 推奨環境 web ブラウザのインストールについて動作 推奨環境以外の新しい web ブラウザを利用していて プレビュー動画試聴に不具合が発生する場合は 下記の手順でブラウザ設定の変更 あるいはダウングレードを行ってください Internet Explorer
動画が視聴できない場合 動作 推奨環境を満たし必要なソフトウェアもインストールしたうえで プレビュー動 画が再生できない場合は以下を確認してください 動作 推奨環境 web ブラウザのインストールについて動作 推奨環境以外の新しい web ブラウザを利用していて プレビュー動画試聴に不具合が発生する場合は 下記の手順でブラウザ設定の変更 あるいはダウングレードを行ってください Internet Explorer