Rでゲノム・トランスクリプトーム解析
|
|
|
- みさえ なつ
- 5 years ago
- Views:
Transcription

1 版 実習用 PC のデスクトップ上に hoge フォルダがあります この中に解析に必要な入力ファイルがあります ネットワーク不具合時は ローカル環境で html ファイルを起動して各自対応してください R で塩基配列解析 : ゲノム解析からトランスクリプトーム解析まで 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
 アグリバイオインフォマティクス教育研究プログラム ( 特別教育研究経費 : 009/4~04/3) アグリバイオインフォマティクス教育研究プログラム")
2 自己紹介 学歴および職歴 少数のスタッフで行っているアグリバイオの活動のみで基本的に手一杯 ここ数年でさらに 研究 << 教育 のヒトに 現在 研究は片手間以下 限界以下のスタッフ数でアグリバイオの本務を行っているため 精神状態をなるべく平静に保つべく 優先順位の低い活動には関与しません 00 年 3 月東京大学 大学院農学生命科学研究科博士課程修了 00 年 4 月産業技術総合研究所 CBRC 003 年 月放射線医学総合研究所 先端遺伝子発現研究センター 005 年 月 ~ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス人材養成プログラム ( 科学技術振興調整費 : 004/0-009/3) アグリバイオインフォマティクス教育研究プログラム ( 特別教育研究経費 : 009/4~04/3) アグリバイオインフォマティクス教育研究プログラム 他大学の学生や社会人も受講できる 希少なバイオインフォ教育プログラム 科目以上の合格者数
3 主な活動 基本スタンスは 優先順位とエフォート 基本独裁 一匹狼 ロビー活動なし 門田教への勧誘なし 信者になっても ( オールフリー派なので w) メリットゼロ 受益者が金と時間をかけずに効率的に学べる教材整備が最優先 東大アグリバイオの大学院講義 ( バイオインフォ全般 ) R を中心としたハンズオン講義 ( 平成 6 年度 ~) 受講人数が多い ( 最大 30 名 ) ので クラウド ( ウェブツール ) 系実習は実質的に不可能 講義補助員 (TA) が数名のみなので Linux 系実習も困難 NBDC/ 東大アグリバイオ /HPCI の NGS ハンズオン講義 (NGS に特化 ) Linux を中心としたハンズオン講義 ( 平成 6 年度 ~) 受講人数は多い ( 最大 7 名 ; おそらくアグリバイオ本体に次ぐ規模 ) が 受講生の意識レベルが高く ( きっちり予習をやるヒトが多数派 ) 環境構築済みノート PC 数 TA 数が充実しているため 本格的な Linux 実習が成立しうる 日本乳酸菌学会誌の NGS 連載 Linux を中心とした自習用教材 ( 平成 6 年度 ~) バクテリア ( 乳酸菌 ) データを 主に Bio-Linux 上で解析するノウハウを提供 第 6 回 (06 年 3 月予定 ) 分以降は DDBJ Pipeline( ウェブツール ) の利用法も紹介 データ取得 インストール 実行に時間がかかるものも 自習なので時間を気にせずにできる ハンズオン講義よりも心穏やか その他 研究 ( 発現変動解析精度向上のためのアルゴリズム開発や評価 ) HPCI 講習会 バイオインフォマティクス実習コースの講師 丸 日だが 上記の主要 3 項目に比べれば心穏やか 3
4 Contents イントロダクション (R で ) 塩基配列解析 アグリバイオ NGS ハンズオン講習会 日本乳酸菌学会の NGS 連載 HPCI 講習会の PC 環境 ゲノム解析 NGS データ解析戦略 DDBJ Pipeline と R の関係 用語説明 de novo アセンブリ実行 および結果を R で解析 塩基配列解析基礎 ( 塩基ごとの出現頻度解析 ) 各種テクニックや注意事項 R コードの解説 塩基配列解析基礎 ( 基本情報取得 ) 塩基配列解析基礎 3( 配列長でフィルタリング ) アノテーション トランスクリプトーム配列 プロモーター配列取得 4
5 (R で ) 塩基配列解析 03 年秋以降の講義資料や連載原稿の PDF を簡単な解説つきで公開 講義資料系は 年以上昔のものは参考程度 ウェブサイトが見づらいとか見栄えに関する要望は無視 ( 優先順位が閾値以下 ) 5

6 (R で ) 塩基配列解析 Linux 系の教材 日本乳酸菌学会誌の NGS 連載 第 4 回 第 5 回 第 6 回は 06 年 3-4 月ごろ公開予定 6

7 (R で ) 塩基配列解析 04 年 4 月刊行の R 本 トランスクリプトーム解析全般の基礎知識的なところは この本の第 章をご覧ください 7
8 (R で ) 塩基配列解析 講習会 講義 講演資料 の PDF 時系列 ( 新 古 ) 順にリストアップ 8

9 アグリバイオ これら 3 科目の講義資料を順番にみていくとよい 科目名 : 農学生命情報科学特論 I 内容 : 公共 DB チェックサム QC 前処理 アセンブリ マッピング RPKM 発現変動など 実施日 : 科目名 : 機能ゲノム学内容 : データ取得 正規化 クラスタリング 発現変動解析 多重比較問題 機能解析など 実施日 : 科目名 : ゲノム情報解析基礎内容 :R の基礎 GC 含量計算や CpG 解析 上流配列解析 R のバージョンの違いなど 実施日 :
10 アグリバイオ 例えば 特論 I の第 4 回講義資料は 講義資料をクリックすればよいが 科目名 : 農学生命情報科学特論 I 内容 : 公共 DB チェックサム QC 前処理 アセンブリ マッピング RPKM 発現変動など 実施日 : 科目名 : 機能ゲノム学内容 : データ取得 正規化 クラスタリング 発現変動解析 多重比較問題 機能解析など 実施日 : 科目名 : ゲノム情報解析基礎内容 :R の基礎 GC 含量計算や CpG 解析 上流配列解析 R のバージョンの違いなど 実施日 :
11 NGS ハンズオン講習会 NGS ハンズオン講習会 のほうが R については基本的なところをきっちり抑えているので や も自習してください

12 NGS ハンズオン講習会 NGS ハンズオン講習会へのリンクもあり
")
13 NGS ハンズオン講習会 大元 (NBDC) のサイトへのリンク NBDC のサイトのほうが見やすいのは 全日程終了から数か月後に整形して公開するから当然です 動画 ( 統合 TV と YouTube) も公開されている 3
14 NGS ハンズオン講習会 R は 05 年 7 月 9-30 日に開催 動画はこちら 4

15 乳酸菌 NGS 連載 NGS 連載関連 主に全体像 原稿およびウェブ資料関係 赤枠の個別の回のところで 原稿中のプログラムへのリンクや コピペ用 Linux コマンドなどを利用可能 5
16 HPCI 講習会の PC 環境 実習用 PC 環境は の手順に従って R および必要なパッケージ のインストールが完了している状態です 自分の PC で復習したい場合は を参考にして自力で環境構築してください 6

17 HPCI 講習会の PC 環境 具体的な順番は R 本体のインストール 各種 R パッケージのインストールです 3 の 基本的な利用法 の習得は HPCI 講習会の枠組みでは必須ではありません 3 7

18 HPCI 講習会 HPCI 講習会の バイオインフォマティクス実習コース の中の一部が門田担当 8
 が門田の担当")
19 HPCI 講習会 具体的には R を使った NGS 解析を基礎から学ぶ のうちの 塩基配列解析 ( 特にゲノム解析とトランスクリプトーム解析部分 ) が門田の担当 9
20 Contents イントロダクション (R で ) 塩基配列解析 アグリバイオ NGS ハンズオン講習会 日本乳酸菌学会の NGS 連載 HPCI 講習会の PC 環境 ゲノム解析 NGS データ解析戦略 DDBJ Pipeline と R の関係 用語説明 de novo アセンブリ実行 および結果を R で解析 塩基配列解析基礎 ( 塩基ごとの出現頻度解析 ) 各種テクニックや注意事項 R コードの解説 塩基配列解析基礎 ( 基本情報取得 ) 塩基配列解析基礎 3( 配列長でフィルタリング ) アノテーション トランスクリプトーム配列 プロモーター配列取得 0
21 NGS データ解析戦略 解析受託企業に外注 :Linux コマンドを知らなくてもよい クラウド ( ウェブツール ):Linux コマンドを知らなくてもよい DDBJ Pipeline (Nagasaki et al., DNA Res., 0: , 03) Illumina BaseSpace Galaxy (Goecks et al., Genome Biol., : R86, 00) Linux コマンドを駆使 ( 旧来型 ) なるべく自力で解析 自分の置かれている環境 予算 ポリシーによっても異なる どの選択肢でも基本正解 R は 主に統計解析部分で使われている Linux コマンドや NGS 解析用プログラムのインストールなどを練習し スパコンを使いこなす NBDC/ 東大アグリバイオ /HPCI の NGS ハンズオン講習会 の方向性
22 DDBJ Pipeline と R 解析受託企業に外注 :Linux コマンドを知らなくてもよい クラウド ( ウェブツール ):Linux コマンドを知らなくてもよい DDBJ Pipeline (Nagasaki et al., DNA Res., 0: , 03) Illumina BaseSpace Galaxy (Goecks et al., Genome Biol., : R86, 00) Linux コマンドを駆使 ( 旧来型 ) なるべく自力で解析 DDBJ Pipelineだけで全てのNGS 解析ができるわけではない Rもまた然り 特にRでは ( 門田の知る限り )de novoアセンブリは不可能 現実を知り うまく使い分けるべし DDBJ Pipeline 上でde novoアセンブリを行った結果の解釈や確認をrで行い 塩基配列解析基礎のスキルがあってよかったと思った実例を紹介 Linux コマンドや NGS 解析用プログラムのインストールなどを練習し スパコンを使いこなす NBDC/ 東大アグリバイオ /HPCI の NGS ハンズオン講習会 の方向性
23 DDBJ Pipeline DDBJ Pipeline では 主に マッピングや de novo アセンブリができる 特に後者ができるのは非常に有難い 3 新規アカウント作成から de novo アセンブリまでの詳細については 乳酸菌連載第 6 回ウェブ資料を参照 3 Nagasaki et al., DNA Res., 0: , 03 3
24 NGS データ 乳酸菌 (Lactobacillus hokkaidonensis LOOC60 T ) ゲノム解読論文 (PMID: ) Illumina MiSeq データ (DRR450) と PacBio データ (DRR04500) を併用することで complete genome を得ることができた という内容 尚 DRR04500 は登録内容の誤りが判明し 06 年 月末に削除され DRR に差し替えられている Tanizawa et al., BMC Genomics, 6: 40, 05 4
 に DRR04500 と DRR450 という ID で登録されていることがわかる Tanizawa et al.")
25 NGS データ Full text リンク先で全文を見られる Availability of supporting data という項目をよく眺めると 生データが DDBJ Sequence Read Archive (DDBJ SRA; 略して DRA) に DRR04500 と DRR450 という ID で登録されていることがわかる Tanizawa et al., BMC Genomics, 6: 40, 05 5
26 NGS データ Genome sequencing and de novo assembly という項目を見ると paired-end で 5,94,60 リードと書いてある 一応公共 DB(DRA) 上で確認する Tanizawa et al., BMC Genomics, 6: 40, 05 6
27 NGS データ Genome sequencing and de novo assembly という項目を見ると paired-end で 5,94,60 リードと書いてある 3DRR0450 という ID のほうは 4,97,30 リードと書いてある 5,94,60 / =,97,30 である ウェブサイト上の数値は single-end としてのリード数と考えれば妥当 3 4 Tanizawa et al., BMC Genomics, 6: 40, 05 7
28 用語 : リード 断片化されたゲノム配列 リードとは Sequencer で読んだ塩基配列のこと 黒矢印の一本一本がリードに相当する single-end の場合 paired-end の場合 8
29 用語 :single-end 断片化されたゲノム配列 断片化された配列の片側のみを読む場合を single-end という この場合は右向き矢印のみ single-endの場合 paired-end の場合 9
30 用語 :paired-end 断片化された配列の両側から読む場合を paired-endという 右向き矢印と3 左向き矢印のリードが読まれることになる それぞれを forward 側リード reverse 側リードなどと呼ぶ 断片化されたゲノム配列 single-end の場合 paired-endの場合 forward 側 3 reverse 側 30
31 用語 :paired-end Illumina MiSeqデータ (DRR450) の場合 forward 側 reverse 側ともに 矢印の長さが 50 bp 矢印の本数( リード数 ) が計 5,94,60 個 ( 約 594 万 ; 片側のみで約 97 万 ) に相当 断片化されたゲノム配列 single-end の場合 paired-end の場合 3
32 DDBJ SRA (DRA) DRA で Illumina MiSeq データ (DRR0450) を概観 Paired-end の FASTQ ファイルをダウンロードする場合は forward 側と 3reverse 側の つに分割されます をクリック 3 3

33 DDBJ SRA (DRA) forward 側 :DRR0450_.fastq.bz reverse 側 :DRR0450_.fastq.bz のような感じ DRA の場合は bzip 圧縮 FASTQ ファイルをダウンロード可能 乳酸菌ゲノム配列決定論文では このデータを入力として de novo アセンブリが行われた Tanizawa et al., BMC Genomics, 6: 40, 05 33
34 用語 : コンティグ 入力 :paired-end FASTQ ファイル ( 通常は )paired-end のリードファイルを入力として de novo アセンブリプログラムを実行した結果として得られる 異なる複数のリードが ( ACGT の切れ目なく ) つなげられたもの contiguous sequence( 連続的な配列 ) という意味 通常 元のリード長よりも長くなる Assembly( コンティグの作成 ) contig contig contig3 contig4 contig5 34
35 用語 :scaffold 入力 :paired-end FASTQ ファイル 得られたコンティグにリードをマップし Assembly( コンティグの作成 ) contig contig contig3 contig4 contig5 Scaffold contig contig contig3 contig4 contig5 35
36 用語 :scaffold 入力 :paired-end FASTQ ファイル 得られたコンティグにリードをマップし ペアの情報を頼りにコンティグ間に N を入れて連結したもの supercontig ともいう scaffold の数は contig の数よりも少なくなる 尚 N を入れた部分を gap という Assembly( コンティグの作成 ) contig contig contig3 contig4 contig5 Scaffold NNNNN NNNN NNNNNN scaffold scaffold 36
37 用語 :gap close 入力 :paired-end FASTQ ファイル 得られた scaffolds にリードをマップし Assembly( コンティグの作成 ) contig contig contig3 contig4 contig5 Scaffold NNNNN NNNN NNNNNN scaffold Gap close scaffold NNNNN NNNN NNNNNN scaffold scaffold 37
38 用語 :gap close 入力 :paired-end FASTQ ファイル 得られた scaffolds にリードをマップし gap 周辺にマップされたリードの塩基で N を置換 gap の N がなくなり 閉じていく (close) ので gap close という ( おそらく ) Assembly( コンティグの作成 ) contig contig contig3 contig4 contig5 Scaffold NNNNN NNNN NNNNNN scaffold Gap close scaffold CA A T C GG G TA A NNNCA TNNC GGNNTA scaffold scaffold 38
39 de novo アセンブリ 入力 :paired-end FASTQ ファイル 最も有名な NGS データ用 de novo ゲノムアセンブリプログラムである Velvet (Zerbino and Birney, Genome Res., 008) は Step までを実行 比較的最近開発された Platanus (Kajitani et al., Genome Res., 04) は Step3 までを実行してくれる Step: Assembly contig contig contig3 contig4 contig5 Step: Scaffold Velvet NNNNN NNNN NNNNNN scaffold scaffold Step3: Gap close Platanus CA A T C GG G TA A NNNCA TNNC GGNNTA scaffold scaffold 39
40 乳酸菌論文は 乳酸菌 (Lactobacillus hokkaidonensis LOOC60 T ) ゲノム解読論文では Illumina MiSeq データ (DRR450) の de novo アセンブリプログラムとして Platanus (ver..) を利用している Tanizawa et al., BMC Genomics, 6: 40, 05 40
41 Contents イントロダクション (R で ) 塩基配列解析 アグリバイオ NGS ハンズオン講習会 日本乳酸菌学会の NGS 連載 HPCI 講習会の PC 環境 ゲノム解析 NGS データ解析戦略 DDBJ Pipeline と R の関係 用語説明 de novo アセンブリ実行 および結果を R で解析 塩基配列解析基礎 ( 塩基ごとの出現頻度解析 ) 各種テクニックや注意事項 R コードの解説 塩基配列解析基礎 ( 基本情報取得 ) 塩基配列解析基礎 3( 配列長でフィルタリング ) アノテーション トランスクリプトーム配列 プロモーター配列取得 4
42 DDBJ Pipeline DDBJ Pipeline では 主に マッピングや de novo アセンブリができる 特に後者ができるのは非常に有難い 3 新規アカウント作成から de novo アセンブリまでの詳細については 乳酸菌連載第 6 回ウェブ資料を参照 ここでは 説明は必要最小限にして R のハンズオンへと移行する 3 Nagasaki et al., DNA Res., 0: , 03 4

43 DDBJ Pipeline で Platanus DDBJ Pipeline のプログラム選択画面 Velvet や Platanus を選択可能 DDBJ Pipeline: Nagasaki et al., DNA Res., 0: , 03 Platanus: Kajitani et al., Genome Res., 4: , 04 43

44 DDBJ Pipeline で Platanus De novo アセンブリの一般的な手順がわかっていれば 赤枠内の Step-3 の説明の意味がなんとなくわかる DDBJ Pipeline は基本的にボタンをポチポチ押していくだけ DDBJ Pipeline: Nagasaki et al., DNA Res., 0: , 03 Platanus: Kajitani et al., Genome Res., 4: , 04 44
45 DDBJ Pipeline で Platanus アセンブリ終了後の画面 Platanus 実行結果ファイル (platanusresult.zip) をダウンロードして解凍したのが Platanus: Kajitani et al., Genome Res., 4: , 04 45
46 DDBJ Pipeline で Platanus アセンブリ終了後の画面 Platanus 実行結果ファイル (platanusresult.zip) をダウンロードして解凍したのが hoge フォルダ中の platanusresult Platanus: Kajitani et al., Genome Res., 4: , 04 46
47 DDBJ Pipeline で Platanus 入力 :paired-end FASTQ ファイル 一般的な de novo アセンブリの手順を知っておけば ファイル名から最終的な結果が 3out_gapClosed.fa だと認識できる Step: Assembly contig contig contig3 contig4 contig5 Step: Scaffold 3 NNNNN NNNN NNNNNN scaffold scaffold Step3: Gap close CA A T C GG G TA A NNNCA TNNC GGNNTA scaffold scaffold 47
48 DDBJ Pipeline と R 解析受託企業に外注 :Linux コマンドを知らなくてもよい クラウド ( ウェブツール ):Linux コマンドを知らなくてもよい DDBJ Pipeline (Nagasaki et al., DNA Res., 0: , 03) Illumina BaseSpace Galaxy (Goecks et al., Genome Biol., : R86, 00) Linux コマンドを駆使 ( 旧来型 ) なるべく自力で解析 DDBJ Pipelineだけで全てのNGS 解析ができるわけではない Rもまた然り 特にRでは ( 門田の知る限り )de novoアセンブリは不可能 現実を知り うまく使い分けるべし DDBJ Pipeline 上でde novoアセンブリを行った結果の解釈や確認をrで行い 塩基配列解析基礎のスキルがあってよかったと思った実例を紹介 Linux コマンドや NGS 解析用プログラムのインストールなどを練習し スパコンを使いこなす NBDC/ 東大アグリバイオ /HPCI の NGS ハンズオン講習会 の方向性 48
49 Contents イントロダクション (R で ) 塩基配列解析 アグリバイオ NGS ハンズオン講習会 日本乳酸菌学会の NGS 連載 HPCI 講習会の PC 環境 ゲノム解析 NGS データ解析戦略 DDBJ Pipeline と R の関係 用語説明 de novo アセンブリ実行 および結果を R で解析 塩基配列解析基礎 ( 塩基ごとの出現頻度解析 ) 各種テクニックや注意事項 R コードの解説 塩基配列解析基礎 ( 基本情報取得 ) 塩基配列解析基礎 3( 配列長でフィルタリング ) アノテーション トランスクリプトーム配列 プロモーター配列取得 49
50 塩基配列解析基礎 入力 :paired-end FASTQ ファイル ( アセンブリ実行結果の )multi-fasta ファイルを読み込んで 塩基ごとの出現頻度解析ができる Step 実行後 (out_contig.fa) は N がなく Step 実行後 (out_scaffold.fa) に N ができて 3Step3 実行後 (out_gapclosed.fa) に N が減るのだろうと妄想できる それを自力で確認することで アルゴリズムの理解を深めることができる Step: Assembly contig contig contig3 contig4 contig5 Step: Scaffold 3 NNNNN NNNN NNNNNN scaffold scaffold Step3: Gap close CA A T C GG G TA A NNNCA TNNC GGNNTA scaffold scaffold 50

multi-")

51 塩基ごとの出現頻度解析 ( アセンブリ実行結果の )multi- FASTA ファイルを読み込んで 塩基ごとの出現頻度解析を行う項目 5
52 塩基ごとの出現頻度解析 例題 7 が Platanus の Step3 実行後のファイル (out_gapclosed.fa) を入力とするものなので そのままコピペできて便利 これを実行します 5
53 塩基ごとの出現頻度解析 つまり Platanus 実行結果ファイル (platanusresult.zip) をダウンロードし 解凍して得られた platanusresult フォルダ中の out_gapclosed.fa を入力として 塩基ごとの出現頻度解析を行う 53

54 R の起動と作業ディレクトリ変更 ファイル ディレクトリの変更 3 デスクトップ hoge - platanusresult を指定する 3 54
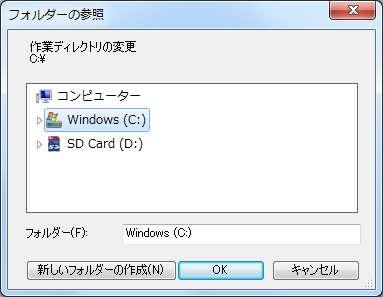
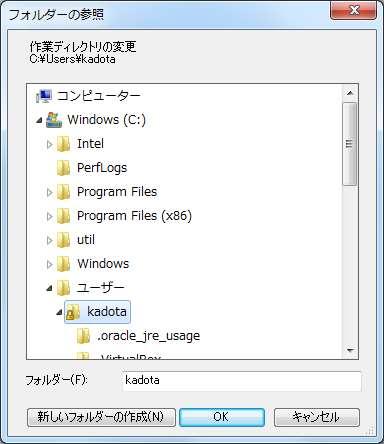
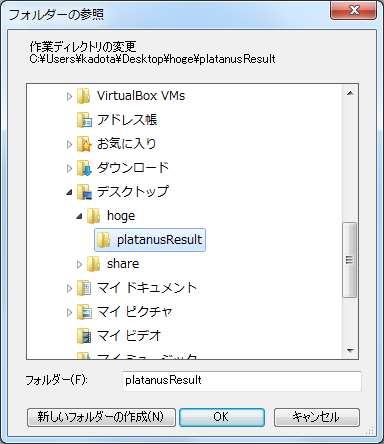
55 作業ディレクトリ変更 ヒトによって若干見栄えは違うだろうが 5-7 が同じになればよい
 と打ち込んで確認 ののように見えていれば")
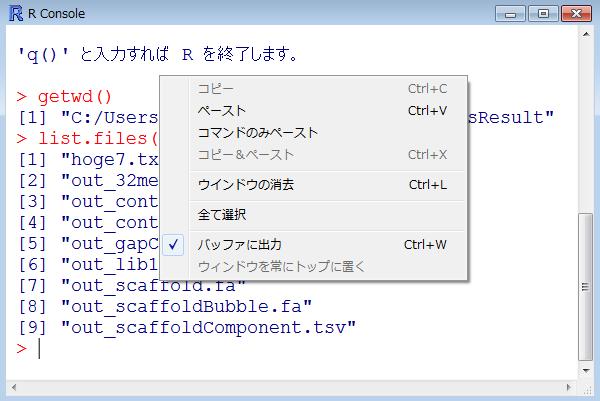
56 getwd() 作業ディレクトリ変更の確認です getwd() と打ち込んで確認 ののように見えていれば OK 56
57 list.files() R 上で 現在の作業ディレクトリ中のファイルを眺めるのが list.files() GUI 画面上で眺めている platanusresult フォルダ中のものと同じものが見えていることがわかる 57

")
58 list.files() 参考 ファイルが存在しないフォルダ上で list.files() とやると character(0) という結果になる 58
59 塩基ごとの出現頻度解析 当たり前ですが 解析したいディレクトリ ( またはフォルダ ) を正しく指定できていなければエラーに遭遇します また 解析したいファイルが存在しない状態でもエラーが出ます 今は 解析したい入力ファイル (out_gapclosed.fa) が R Console 画面上でも 3 見えているのでエラーなく動くはずです 3 59


60 基本はコピペ 一連のコマンド群をコピーして R Console 画面上でペースト Windows のヒトは CTRL と ALT キーを押しながらコードの枠内で左クリックすると 全選択できます トリプルクリックでも OK Macintosh はよくわかりません 60

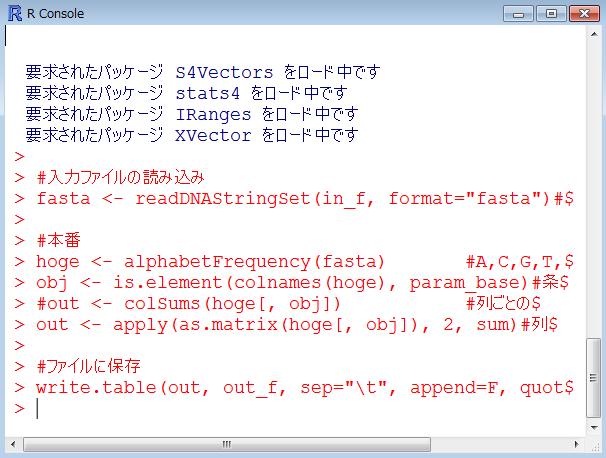
61 途中経過と終了後 コピペ直後と 実行後の状態 エラーなく実行できたときはこんな感じになります 一見何も変化がないように見えますが 6

62 結果の解説 解析結果 ( 塩基ごとの出現頻度情報 ) は hoge7.txt というファイルに保存されている list.files() とやると 確かに自分が出力ファイル名として指定した 3hoge7.txt が存在することがわかる 6
 は手の届く場所 ( つまり作業ディレクトリ内 )")
 はただの確認用 4")
63 結果の解説 もちろん出力ファイル (hoge7.txt) は手の届く場所 ( つまり作業ディレクトリ内 ) にある getwd() や 3 現在時刻を表示する date() はただの確認用 4 エクセルで眺めるとこんな感じ

64 R 上で眺める 赤枠程度の情報量なら エクセルなどをわざわざ開くまでもなく R 上で眺めればよい 例えば ここでは 出力ファイル名を out_f というオブジェクト名で取り扱っている 3out_f と打てば対応関係がわかる 3 64

65 R 上で眺める R コードの最後の部分が ファイルに保存するところ out_f に書き込んでいるのは out というオブジェクトの情報 3out の中身を見れば hoge7.txt と同じ情報を得られる 3 65
 は 入力ファイル (out_gapclosed.")
66 sum で総塩基数を得る out オブジェクトは 数値ベクトル sum は 数値ベクトルの総和を計算する関数 out に対して実行した結果 (,356,09) は 入力ファイル (out_gapclosed.fa) の総塩基数を調べていることと同義 66
67 sum で総塩基数を得る DDBJ Pipeline 実行結果画面上の数値と同じ 入力ファイル (out_gapclosed.fa) は DDBJ Pipeline 上で Platanus という de novo アセンブリプログラムを実行した結果だったことを思い出そう 67
68 目的をおさらい 入力 :paired-end FASTQ ファイル ( アセンブリ実行結果の )multi-fasta ファイルを読み込んで 塩基ごとの出現頻度解析ができる Step 実行後 (out_contig.fa) は N がなく Step 実行後 (out_scaffold.fa) に N ができて 3Step3 実行後 (out_gapclosed.fa) に N が減るのだろうと妄想できる それを自力で確認することで アルゴリズムの理解を深めることができる Step: Assembly contig contig contig3 contig4 contig5 Step: Scaffold 3 NNNNN NNNN NNNNNN scaffold scaffold Step3: Gap close CA A T C GG G TA A NNNCA TNNC GGNNTA scaffold scaffold 68
69 目的をおさらい 入力 :paired-end FASTQ ファイル ( アセンブリ実行結果の )multi-fasta ファイルを読み込んで 塩基ごとの出現頻度解析ができる Step 実行後 (out_contig.fa) は N がなく Step 実行後 (out_scaffold.fa) に N ができて 3Step3 実行後 (out_gapclosed.fa) に N が減るのだろうと妄想できる それを自力で確認することで アルゴリズムの理解を深めることができる を調べるにはどうすればいいか? Step: Assembly contig contig contig3 contig4 contig5 Step: Scaffold NNNNN NNNN NNNNNN scaffold scaffold Step3: Gap close CA A T C GG G TA A NNNCA TNNC GGNNTA scaffold scaffold 69
70 Contents イントロダクション (R で ) 塩基配列解析 アグリバイオ NGS ハンズオン講習会 日本乳酸菌学会の NGS 連載 HPCI 講習会の PC 環境 ゲノム解析 NGS データ解析戦略 DDBJ Pipeline と R の関係 用語説明 de novo アセンブリ実行 および結果を R で解析 塩基配列解析基礎 ( 塩基ごとの出現頻度解析 ) 各種テクニックや注意事項 R コードの解説 塩基配列解析基礎 ( 基本情報取得 ) 塩基配列解析基礎 3( 配列長でフィルタリング ) アノテーション トランスクリプトーム配列 プロモーター配列取得 70
71 入力ファイルを変更 テンプレートの out_gapclosed.fa を out_scaffold.fa に変更すればよい 7
 に例題をコピペし")
72 入力ファイルを変更 適当なテキストエディタ ( ここでは EmEditor) に例題をコピペし 必要最小限の変更を施したところ 7
73 変更後のコードをコピペ コピペ 73
74 ありがちなミス これはエラーメッセージです w エラーの理由は 出力予定ファイル (hoge7.txt) を開くことができない というもの Permission denied( 権限が与えられていない ) は アク禁 みたいなものです Tips: ワードパッド や メモ帳 で開く分にはエラーは出ないようです 74
75 ありがちなミス エラーの原因は エクセルで hoge7.txt を開いているから 閉じて再実行すればエラーは出なくなる 75

76 再実行 エクセルを閉じて再実行した結果 エラーは出ていないことがわかる out オブジェクトの中身を見ると 確かに N がある! 76
 のイメージは のような感じなので N が 49 個あったという結果は合理的 Step: Assembly contig contig contig3 contig4 contig5 Step: Scaffold NNNNN NNNN NNNNNN scaffold")
77 納得できる結果 入力 :paired-end FASTQ ファイル 入力ファイル (out_scaffold.fa) のイメージは のような感じなので N が 49 個あったという結果は合理的 Step: Assembly contig contig contig3 contig4 contig5 Step: Scaffold NNNNN NNNN NNNNNN scaffold scaffold Step3: Gap close CA A T C GG G TA A NNNCA TNNC GGNNTA scaffold scaffold 77
78 ありがちなミス 最終行の部分で 改行をキチンと含めないとハマる 78

79 ありがちなミス 最終行の部分で 改行をキチンと含めないと 最後の write.table 関数部分が実行されない つまりファイルが作成されません 79
80 実際の利用時は hoge フォルダ直下にある rcode.txt のような 無駄なコメントを除いてスリムにした一連のスクリプトを作成しておき 一気にコピペ 80
81 一気に結果を得る hoge フォルダ直下にある rcode.txt のような 無駄なコメントを除いてスリムにした一連のスクリプトを作成しておき 一気にコピペ コピペ後に自分が指定した出力ファイルができていることを確認 8
82 結果のまとめ result_step*.txt の結果をまとめたものが 8
83 結果の解釈 入力 :paired-end FASTQ ファイル Step 実行後は N が 0 Step 実行後に N が 49 個生成されたということは いくつかの contigs がまとめられて scaffolds になったのだろう 3 Step3 で N が 0 個になったのはおそらくたまたまうまくいっただけ 49 個よりも減ったということが重要で gap close がうまく機能したと判断できる Step: Assembly contig contig contig3 contig4 contig5 Step: Scaffold 3 NNNNN NNNN NNNNNN scaffold scaffold Step3: Gap close CA A T C GG G TA A NNNCA TNNC GGNNTA scaffold scaffold 83
84 コード内部の理解は重要 入力 :paired-end FASTQ ファイル R を使うことで アセンブリプログラムの内部挙動の把握や理解ができる 他の例は 配列数 (contig 数や scaffold 数と書くと説明しづらいので配列数に統一 ) 配列数は Step Step で減り Step Step3 では不変だろうと予想 Step: Assembly contig contig contig3 contig4 contig5 Step: Scaffold NNNNN NNNN NNNNNN scaffold scaffold Step3: Gap close CA A T C GG G TA A NNNCA TNNC GGNNTA scaffold scaffold 84
85 Contents イントロダクション (R で ) 塩基配列解析 アグリバイオ NGS ハンズオン講習会 日本乳酸菌学会の NGS 連載 HPCI 講習会の PC 環境 ゲノム解析 NGS データ解析戦略 DDBJ Pipeline と R の関係 用語説明 de novo アセンブリ実行 および結果を R で解析 塩基配列解析基礎 ( 塩基ごとの出現頻度解析 ) 各種テクニックや注意事項 R コードの解説 塩基配列解析基礎 ( 基本情報取得 ) 塩基配列解析基礎 3( 配列長でフィルタリング ) アノテーション トランスクリプトーム配列 プロモーター配列取得 85
 実行時に pattern オプションをつけて任意の文字列を含むもののみ表示させることが可能 ここでは out_")
86 R コードの解説 配列数の把握の仕方 の前に Tips list.files() 実行時に pattern オプションをつけて任意の文字列を含むもののみ表示させることが可能 ここでは out_ という文字列を含むもの ( ファイル ) のみ表示させている 入力ファイルの存在確認 86
87 R コードの解説 配列数の把握の仕方 赤枠部分をコピペ 87
88 R コードの解説 4 Biostrings という R パッケージを library 関数で読み込んで Biostrings パッケージが提供する関数群を利用可能な状態にする (Biostrings が提供する )readdnastringset 関数を用いて 3FASTA 形式の 4 入力ファイルを読み込んだ結果を 5fasta というオブジェクト ( もの という理解でよい ) に格納

89 Tips: 配列数 fasta オブジェクトの中身を表示 ( ここでの目的の ) 配列数は 7 個 スカラー値として配列数情報のみ取り出したい場合は 3 ベクトルの要素数を調べる length 関数を利用する 3 89
90 答え合わせ DDBJ Pipeline 実行結果の数値 (7 個 ) と同じことがわかります 最長の配列 (Maximum contig size; 57,78 bp) と最短の配列 (Minimum contig size; 0 bp) も R 上で把握できます 90
fasta オブジェクト中の width")
91 Tips: 配列長 配列長の情報は (DNAStringSet という形式で保持されている )fasta オブジェクト中の width 列の位置に相当する 9
 の配列数なら パッと見で 最長と 3")
92 Tips: 配列長 配列長情報は width(fasta) とやることで 数値ベクトルとして取り出すことができる この程度 (7 個 ) の配列数なら パッと見で 最長と 3 最短のものを確認できるが 3 9
, digits=6) とすれば の 57700 が正しく 5778 と表示されるようになる")
93 Tips: 配列長 ベクトル演算の基本関数を駆使して全貌を把握する 上矢印キーを 回押して 以前打ち込んだコマンドを出すなど 上下左右の矢印キーを駆使して効率的に打ち込むべし や の数値は つ前のスライドには存在しないが これは 3summary 関数実行結果として表示させる有効数字のデフォルトが 4 桁だから summary(width(fasta), digits=6) とすれば の が正しく 5778 と表示されるようになる 3 93
 ベクトルとして取り扱うことができる ここでは 3:4 という指定を行って 最初の 4 個分のみ表示させている 3 94")
94 Tips:description 部分 description 行部分は names という関数を用いることで ( 文字列 ) ベクトルとして取り扱うことができる ここでは 3:4 という指定を行って 最初の 4 個分のみ表示させている 3 94
95 Tips: 塩基配列部分 但し このノリは塩基配列部分には通用しない w seq という関数は別の意味を持つこと fasta オブジェクトの主要な中身がこの塩基配列情報であるため と理解すればよい 95

96 Tips: 塩基配列部分 どうしても文字列ベクトルなどで取り出したい場合は as.character 関数を使うが DNAStringSet 形式の fasta オブジェクトをそのまま用いて各種塩基配列解析を行うのが通常のやり方 96
97 Contents イントロダクション (R で ) 塩基配列解析 アグリバイオ NGS ハンズオン講習会 日本乳酸菌学会の NGS 連載 HPCI 講習会の PC 環境 ゲノム解析 NGS データ解析戦略 DDBJ Pipeline と R の関係 用語説明 de novo アセンブリ実行 および結果を R で解析 塩基配列解析基礎 ( 塩基ごとの出現頻度解析 ) 各種テクニックや注意事項 R コードの解説 塩基配列解析基礎 ( 基本情報取得 ) 塩基配列解析基礎 3( 配列長でフィルタリング ) アノテーション トランスクリプトーム配列 プロモーター配列取得 97
 4K (G or T) など 門田は シロイヌナズナのゲノム配列で ACGTN")
98 alphabetfrequency 塩基ごとの出現頻度解析の中核となっている関数は alphabetfrequency 実行結果である hoge の中身は数値行列 この段階で塩基配列解析から数値解析に切り替わる 塩基の種類には多型がある 例えば 3M (A or C) 4K (G or T) など 門田は シロイヌナズナのゲノム配列で ACGTN 以外のものを見た記憶あり

99 dim dim 関数で行数と列数を把握 alphabetfrequency は 配列ごとに結果を返しているので 7 行からなると解釈 8 列であることから 塩基の種類数は 8 個と解釈 3 行列の一部要素の取り出し例 3 99
 を行っている 行列 hoge の列名 (column names) からなるベクトルの中から 3param_base で指定された要素が存在する場所を TRUE")
100 is.element は 出現頻度情報を得たい塩基の種類を指定するところ ほとんどの場合 ACGTN のみで事足りるので このようにしている is.element 関数は条件判定 ( 集合演算 ) を行っている 行列 hoge の列名 (column names) からなるベクトルの中から 3param_base で指定された要素が存在する場所を TRUE そうでないところを FALSE と評価するのが 4is.element 関数
![条件を満たす列のみ 行列の subsetting は [ 行, 列 ] で指定する [, 列 ] で列のみの指定 [ 行, ] で行のみの指定となる hoge[, obj] は obj ベクトルの TRUE](/docs-images/88/115135629/images/101-1.jpg "となっている列の位置のみ取り出すことに相当する hoge[:, ] で hoge 行列の最初の 行分のみ表示 3hoge[:, obj] の合わせ技で さらに param_base")
101 条件を満たす列のみ 行列の subsetting は [ 行, 列 ] で指定する [, 列 ] で列のみの指定 [ 行, ] で行のみの指定となる hoge[, obj] は obj ベクトルの TRUE となっている列の位置のみ取り出すことに相当する hoge[:, ] で hoge 行列の最初の 行分のみ表示 3hoge[:, obj] の合わせ技で さらに param_base で指定した塩基のみを出力できるようになる 3 0
102 colsums alphabetfrequency 実行結果は 配列ごとに各塩基の出現頻度を計算している そのため hoge は 7 行分の要素からなる colsums は 行列データを入力として 列ごとに総和 (column sum) を計算する関数 colsums を適用することで 配列ごとではなくファイル全体の出現頻度を得ることができる ( 今得たい情報はこれ ) 0
103 colsums は最初の 行分のみで列ごとの総和を計算する場合 ではエラーとなっている colsums の入力として与えている hoge[, obj] は 最初の 行分のみからなる つまり 入力が 次元の行列データではなく 次元のベクトルになってしまっているため 3 行頭に # をつけており 実際にはこのコードは動作していない 3 03
104 apply が一般的かも このコードで実際に動かしているのは apply 関数を用いるほう 結果は colsums と同じ おそらく行列演算で行ごとや列ごとに何かを行うときには 一般に apply 関数を用いるので 一応示した 04
105 apply の説明 apply は 入力データに対して 列ごと ( 行ごとの場合はここを にする ) に 3 総和を計算する sum 関数を適用する みたいな指定を行う colsums だと sum を計算することしかできないが apply の場合は 3 のところの関数名を mean, median, max などいろいろ自在に変更できる 3 05
106 as.matrix 実はこの入力ファイルの場合は as.matrix という関数を つけなくても 3 つけたときと同じ結果が得られる つけている理由は apply(as.matrix( ),, sum) などとして行ごとに sum 関数を適用したいときに 配列数が複数の場合でも単数の場合でも統一的にエラーなく処理できたという記憶があったから 3 06
107 as.matrix 挙動の違いは 入力データの行列が 行しかない ( 配列数が つの ) 場合に出てくる 複数行からなる ( 配列数が つ以上の ) 場合と比べればエラーメッセージの違いがわかります 07
108 思考停止するべからず as.matrix をつけてエラーメッセージが出てないからといって これが正しいわけではないことに気づこう 08

109 思考停止するべからず の実行結果である 050 という数値は 単純に 番目の配列の長さ 今調べたいのは塩基ごとの出現頻度情報なので 3 が正解! 3 09
110 思考停止するべからず 少なくともこのサンプルコードは 配列数が つしかない場合にはうまく動かないことが既知 欲しい結果が ( この場合は ) 数値ベクトルになっていない段階でおかしいと思えるようになりましょう 一般論としては 得られる結果をイメージし 特にイメージと異なる場合に疑いの目で結果を眺めよう 0
111 R コードの解説 赤下線のように沢山のオプションを駆使している sep= t は区切り文字を指定するオプション t はタブ区切りの意味 row.names=t は 行の名前 (row names) を TRUE にせよという意味 ここが T になると 3 赤枠部分の情報が追加される FALSE にすると この列は消える 4 col.names=f は col.names=t にしたときに無意味なヘッダー行が含まれるのが嫌だったのでこうしているだけ 3 4
112 目的をおさらい 入力 :paired-end FASTQ ファイル 配列数は Step Step で減り Step Step3 では不変だろうと予想 Step: Assembly contig contig contig3 contig4 contig5 Step: Scaffold NNNNN NNNN NNNNNN scaffold scaffold Step3: Gap close CA A T C GG G TA A NNNCA TNNC GGNNTA scaffold scaffold

113 目的をおさらい 3 配列数は Step Step で減り Step Step3 では不変だろうと予想 3hoge フォルダ直下の rcode.txt は 配列数をカウントする必要最小限のコード で予想通りの結果 3
114 Contents イントロダクション (R で ) 塩基配列解析 アグリバイオ NGS ハンズオン講習会 日本乳酸菌学会の NGS 連載 HPCI 講習会の PC 環境 ゲノム解析 NGS データ解析戦略 DDBJ Pipeline と R の関係 用語説明 de novo アセンブリ実行 および結果を R で解析 塩基配列解析基礎 ( 塩基ごとの出現頻度解析 ) 各種テクニックや注意事項 R コードの解説 塩基配列解析基礎 ( 基本情報取得 ) 塩基配列解析基礎 3( 配列長でフィルタリング ) アノテーション トランスクリプトーム配列 プロモーター配列取得 4
115 塩基配列解析基礎 の情報を一気に得る項目もあります 5

116 塩基配列解析基礎 FASTA 形式ファイルを読み込んで各種情報を得る項目 6
117 塩基配列解析基礎 rcode3.txt 3 例題の 入力ファイル名部分を out_gapclosed.fa に変更した 3rcode3.txt をコピペで実行 7
118 塩基配列解析基礎 rcode3.txt 出力ファイル (hoge.txt) を開かずに write.table 関数で書きだしている tmp の中身を表示 8
119 塩基配列解析基礎 rcode3.txt DDBJ Pipeline 上の Platanus 実行結果と完全一致 GC 含量情報なども得られる 9
120 塩基配列解析基礎 rcode3.txt 配列数の算出法 length(fasta) や 最短配列長 min(width(fasta)) も前のスライドで解説したものと同じです 0
121 塩基配列解析基礎 3 このアセンブル結果の最短配列長は 0 bp 通常 アセンブル結果ファイルから一定の配列長 ( 例 :300 bp) 未満のものは除去される



122 塩基配列解析基礎 3 FASTA 形式ファイルを読み込んで 指定した配列長以上のもののみ残して FASTA 形式ファイルで出力する項目 例題 5 は out_gapclosed.fa を読み込んで 300 bp 以上の配列のみ hoge5.fasta ファイルに保存するスクリプト

123 塩基配列解析基礎 3 赤枠部分をコピペ 入力ファイルを読み込んだ直後の fasta オブジェクトは 37 個の配列からなる 赤下線で見えているものが 300 bp 未満なのでフィルタリングされる 3 3

124 塩基配列解析基礎 3 width(fasta) は配列長情報からなる数値ベクトル 300 bp という閾値情報からなる param_length で条件判定した結果が obj に格納されている 4

125 塩基配列解析基礎 3 param_length 以上 (>=) という条件を満たすものが TRUE そうでないものが FALSE 5
")
126 塩基配列解析基礎 3 オリジナルの 7 配列からなる fasta オブジェクトの中から obj が TRUE となる (300 bp 以上の ) 配列は 5 個 6
127 塩基配列解析基礎 3 こういう上書きはアリです もちろん fasta みたいな別名にしてもいいが ヒトゲノム配列などを取り扱うときにはノート PC レベルではメモリ的に厳しくなります 7
128 塩基配列解析基礎 3 writexstringset 関数を使えば fastaオブジェクトの中身を指定したファイルに書きだすことができる のXStringSetのXは 何でもよいみたいな意味 fastaがdnastringssetという形式で格納されていること アミノ酸配列 (Amino Acids) を格納する形式として AAStringSetという形式が存在することから それらを同じ関数で統一的に取り扱えるようにするため 8

129 塩基配列解析基礎 3 出力ファイルは FASTA 形式で保存した hoge5.fasta 行あたりの塩基数を 50 個に指定している 9
130 Contents イントロダクション (R で ) 塩基配列解析 アグリバイオ NGS ハンズオン講習会 日本乳酸菌学会の NGS 連載 HPCI 講習会の PC 環境 ゲノム解析 NGS データ解析戦略 DDBJ Pipeline と R の関係 用語説明 de novo アセンブリ実行 および結果を R で解析 塩基配列解析基礎 ( 塩基ごとの出現頻度解析 ) 各種テクニックや注意事項 R コードの解説 塩基配列解析基礎 ( 基本情報取得 ) 塩基配列解析基礎 3( 配列長でフィルタリング ) アノテーション トランスクリプトーム配列 プロモーター配列取得 30
 があり どんな Gene Ontology ID や KEGG Pathway 上に存在するかなどを得る作業 広範囲 KEGG 系 3 バクテリアに特化")
131 アノテーション アノテーション ( 遺伝子注釈付け ) は アセンブル後の配列を入力として与え どこ ( 座標 ) にどんな遺伝子 (gene symbols; gene names; products) があり どんな Gene Ontology ID や KEGG Pathway 上に存在するかなどを得る作業 広範囲 KEGG 系 3 バクテリアに特化 などいろいろあります 3 3


132 アノテーション アノテーションファイルの形式は GFF/GTF が有名 3
133 GFF/GTF 形式ファイルの例 GFF3 形式 ( シロイヌナズナ ; TAIR0_GFF3_genes.gff) 他に refflat 形式など様々なファイル形式が存在します GTF 形式 ( ゼブラフィッシュ ; Danio_rerio.Zv9.75.gtf) 33
134 GFF の読み込み 読み込み段階でコケる 読み込みはうまくいったが その後の解析段階でコケるなど Linux 上での解析同様 一筋縄ではいきません 過去の受講生など多方面からの情報提供のおかげでだいぶ分かってきました 34
135 GFF の読み込み 例題 7 ここで用いている GFF 形式の入力ファイルは 3 から取得しました 3 をクリックしたつもりでよい w 3 35

136 Ensembl 解説 GFF ファイルはここから取得 の gzip 圧縮ファイルをダウンロードして解凍したものが入力ファイル 3 のあたりがバージョン番号 概ね月単位でバージョン番号が上がっていく 3 36
137 Ensembl 解説 で このゲノムの全貌をある程度把握可能 原著論文の情報なども合わせることで chromosome and plasmids 環状ゲノムであることも認識可能 3 でゲノム配列も取得できる 3 Tanizawa et al., BMC Genomics, 6: 40, 05 37

138 Ensembl 解説 いろんなものがあって私はよくわかりませんが GFF ファイルと一緒に取り扱いたいときには GFF ファイルと似た名前の を採用します Tanizawa et al., BMC Genomics, 6: 40, 05 38


139 GFF の読み込み 例題 7 が読み込みの基本形 GenomicFeatures というパッケージが提供する 3makeTxDbFromGFF 関数を用いて GFF ファイルを読み込んで TxDb という独特の形式で取り扱えるようにする 入力ファイルは 4 hoge L.hokkaidonensis 中にある
140 GFF の読み込み 入力ファイルが GFF ver.3 という形式になっていないみたいな警告メッセージが出ているが 読み込んだ後の txdb オブジェクトは大丈夫そうだ たぶん 40
141 GFF の読み込み 若干自信がないのは GFF ファイル読み込み後の で見えている数値と Ensembl ウェブサイト上で見られる数値が一致していないことに由来,344 や,4 はプラスミドを含むものなのか詳細は不明 4
142 Contents イントロダクション (R で ) 塩基配列解析 アグリバイオ NGS ハンズオン講習会 日本乳酸菌学会の NGS 連載 HPCI 講習会の PC 環境 ゲノム解析 NGS データ解析戦略 DDBJ Pipeline と R の関係 用語説明 de novo アセンブリ実行 および結果を R で解析 塩基配列解析基礎 ( 塩基ごとの出現頻度解析 ) 各種テクニックや注意事項 R コードの解説 塩基配列解析基礎 ( 基本情報取得 ) 塩基配列解析基礎 3( 配列長でフィルタリング ) アノテーション トランスクリプトーム配列 プロモーター配列取得 4
 と")
 を同時に読み込むことで 例えば")

143 転写物配列取得 multi-fasta ファイル ( ゲノム配列情報 ) と GFF ファイル ( アノテーション情報 ) を同時に読み込むことで 例えば トランスクリプトーム ( 転写物 ) 配列情報を一気に取得することも可能 例題 5 43
144 転写物配列取得 は GFF ファイル情報を保持した txdb オブジェクトから transcripts という関数を用いて抽出したい転写物の座標情報を取得した結果を hoge に保存している 44
145 転写物配列取得 GFF ファイルの見方がよくわかっていなくても うまく読み込めているらしいことはわかる 45
getseq 関数を用いて取得 4(Rsamtools パッケージが提供する )FaFile 関数は getseq 関数利用時に必要なおまじない 4 4 3")
146 転写物配列取得 in_f で指定したゲノム配列情報はここで登場 ゲノム配列から で指定した座標情報の塩基配列を 3(Biostrings パッケージが提供する )getseq 関数を用いて取得 4(Rsamtools パッケージが提供する )FaFile 関数は getseq 関数利用時に必要なおまじない

147 転写物配列取得 getseq 実行後の fasta オブジェクトが 欲しいトランスクリプトーム配列情報ではあるが 47
148 転写物配列取得 の fasta オブジェクトをそのまま FASTA 形式で保存すると で見えているがままの description 情報が書きだされる つまり すべて Chromosome になってしまう 48

149 転写物配列取得 赤枠部分で行っているのは description 部分の記述内容を Chromosome_start_end としてどこの座標由来の塩基配列かがわかるようにしている paste は 文字列を sep オプションで指定した文字を間に挟んで連結する関数 3 の例をみれば挙動がわかると期待 3 49

150 転写物配列取得 description 部分が変わっていることがわかる これを眺めるだけで 出力ファイルをみなくてもうまくいっていると判断できる ( と油断していると時々落とし穴があるので注意 ) 50
151 Contents イントロダクション (R で ) 塩基配列解析 アグリバイオ NGS ハンズオン講習会 日本乳酸菌学会の NGS 連載 HPCI 講習会の PC 環境 ゲノム解析 NGS データ解析戦略 DDBJ Pipeline と R の関係 用語説明 de novo アセンブリ実行 および結果を R で解析 塩基配列解析基礎 ( 塩基ごとの出現頻度解析 ) 各種テクニックや注意事項 R コードの解説 塩基配列解析基礎 ( 基本情報取得 ) 塩基配列解析基礎 3( 配列長でフィルタリング ) アノテーション トランスクリプトーム配列 プロモーター配列取得 5
152 プロモーター配列取得 基本的には プロモーター配列取得もトランスクリプトームのときと同じノリ 例題 0 5

153 プロモーター配列取得 例題 0 は 転写開始点上流 00 bp 下流 0 bp の領域を取得するコード はその領域情報 これだけだと 3 確かに 0 bp 分の領域になっていることの確認しかできない 3 53
 で strand")
154 プロモーター配列取得 例題 0 は 転写開始点上流 00 bp 下流 0 bp の領域を取得するコード 元となっている転写開始点情報を transcripts(txdb) で strand 情報も含めて比較するとよくわかる 54

155 プロモーター配列取得 例題 0 は 転写開始点上流 00 bp 下流 0 bp の領域を取得するコード 最後まで実行した結果 転写開始点上流 00 bp 下流 0 bp の領域を取得するコードなので 配列長が全て 0 bp になっており妥当 55
156 失敗例 例題 は 転写開始点上流 00 bp 下流 0 bp の領域を取得するコード 例題 0 との違いは 上流の塩基配列数のみ 56

157 失敗例 例題 は 転写開始点上流 00 bp 下流 0 bp の領域を取得するコード 例題 0 との違いは 上流の塩基配列数のみ 57

158 失敗例 例題 は 転写開始点上流 00 bp 下流 0 bp の領域を取得するコード 例題 0 との違いは 上流の塩基配列数のみ 58

159 失敗例 例題 は 転写開始点上流 00 bp 下流 0 bp の領域を取得するコード 例題 0 との違いは 上流の塩基配列数のみ hoge は取得したいプロモーター配列の座標情報 3getSeq を実行するとエラーが出る 3 59
160 思考停止するべからず 例題 は 転写開始点上流 00 bp 下流 0 bp の領域を取得するコード 例題 0 との違いは 上流の塩基配列数のみ hoge は取得したいプロモーター配列の座標情報 3getSeq を実行するとエラーが出る 4 fasta と打つと何か出力されるがうまく取れているわけではない!
161 思考停止するべからず 3 例題 は 転写開始点上流 00 bp 下流 0 bp の領域を取得するコード この fasta オブジェクトの中身は 3 この R Console 画面上で以前に行っていた例題 0( 転写開始点上流 00 bp 下流 0 bp の領域を取得するコード ) 実行時に作成されたものが残っているだけである その証拠に 4 ここが 0 4 6


162 大事な計算時は Rを再起動し 真っ新な状態でコピペするのが一番スッキリ 私はいつもコレ 普段は いいえ を押して作業スペースを保存せずに終了させるが ここでは3キャンセルにしておく 3 6
 63")
163 オブジェクトの表示 現在利用可能なオブジェクトの表示は ls() 63
 全オブジェクトの消去は rm(list =")
164 オブジェクトの消去 ( この R Console 画面上で利用可能な ) 全オブジェクトの消去は rm(list = ls()) 64
165 Contents イントロダクション (R で ) 塩基配列解析 アグリバイオ NGS ハンズオン講習会 日本乳酸菌学会の NGS 連載 HPCI 講習会の PC 環境 ゲノム解析 NGS データ解析戦略 DDBJ Pipeline と R の関係 用語説明 de novo アセンブリ実行 および結果を R で解析 塩基配列解析基礎 ( 塩基ごとの出現頻度解析 ) 各種テクニックや注意事項 R コードの解説 塩基配列解析基礎 ( 基本情報取得 ) 塩基配列解析基礎 3( 配列長でフィルタリング ) アノテーション トランスクリプトーム配列 プロモーター配列取得 65

166 例題 再実行 Tips ディレクトリの変更は setwd でも可能 お約束のコマンドは のような任意のファイルに書き込んでおいて R 起動直後に無条件でコピペしておく ( のが門田の習慣 ) 66

167 例題 再実行 例題 再実行結果 getseq 実行部分でコケるところまでは一緒 3 以降は fasta オブジェクトがないのでそれを用いる部分は軒並みエラー祭りになっていることがわかる 重要なのは エラーの原因を正確に把握すること 3 67
168 エラー原因解説 エラーを把握すべく 欲しい領域 hoge と 3getSeq 実行部分を再度表示 3 68
169 エラー原因解説 エラーの原因は に書かれているように 6 番目のレコード (Chromosome: ) が切り捨てられている (truncated) というもの これは 3 最後の転写開始点 (77853 番目の塩基 ) の上流 00 bp から下流 0 bp の領域に相当する 3 69
 を取得可能 3 で取得しようとしていた領域の一部が存在しないことが原因である")
170 エラー原因解説 seqinfo 関数を使うことで in_f で指定したファイルの配列長情報 (77985 bp) を取得可能 3 で取得しようとしていた領域の一部が存在しないことが原因である 3 70
 今問題となっている の領域が除去")
171 例題 が推奨コード 例題 は 取得予定の座標が存在するかどうかを判定し 存在しないものをフィルタリングする部分を追加したコード ( 甲斐政親氏提供 ) 今問題となっている の領域が除去 (filter out) されればよい それを行ってくれる 7
172 例題 が推奨コード 3 が取得予定の座標が存在するかどうかを判定し 存在しないものをフィルタリングする部分 ( 甲斐政親氏提供 ) 3 7

173 例題 が推奨コード 赤枠のフィルタリング実行後の状態 エラーの原因であった 6 番目の領域がなくなっていることがわかる 73

174 例題 が推奨コード 例題 を最後まで実行した結果 74
175 例題は多数 つの項目内にも多数の例題があります うまく動かないままわかってて放置してあるもの 気づいていないもの 作成当時はうまく動いていたが R 本体のバージョンが上がってからうまく動かなくなっているもの 条件判定が不十分なものなど 玉石混交です 75
176 Contents トランスクリプトーム解析 イントロダクション : 簡単な原理 基本イメージ 様々な解析目的 解析データ : 乳酸菌 (L. casei A) QuasRでマッピング ( 基礎 ): コード各部の説明と結果の解釈 QuasRでマッピング ( 応用 ): オプションを指定して実行 カウント情報取得, サンプル間クラスタリング (TCC) 発現変動解析 (TCC) M-A plot モデル 分布 統計的手法 3 群間比較 (TCCによるANOVA 的な解析 ) 遺伝子間クラスタリング (MBCluster.Seq) 3 群間比較 (TCCによるANOVA 的な解析 + MBCluster.Seqでのパターン分類 ) 76
177 トランスクリプトーム解析 ある状態のあるサンプル ( 例 : 目 ) のあるゲノムの領域 遺伝子 遺伝子 遺伝子 3 遺伝子 4 遺伝子全体 ( ゲノム ) どの染色体上のどの領域にどの遺伝子があるかは調べる個体 ( 例 : ヒト ) が同じなら不変 ( 目だろうが心臓だろうが ) 働いている RNA の種類や量を調べるのが目的 ヒト AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA 転写物全体 ( トランスクリプトーム ) mrna 遺伝子 は沢山転写されている ( 発現している ) 遺伝子 4 はごくわずかしか転写されてない 77
178 トランスクリプトーム解析 ある状態のあるサンプル ( 例 : 目 ) のあるゲノムの領域 遺伝子 遺伝子 遺伝子 3 遺伝子 4 遺伝子全体 ( ゲノム ) どの染色体上のどの領域にどの遺伝子があるかは調べる個体 ( 例 : ヒト ) が同じなら不変 ( 目だろうが心臓だろうが ) 働いているRNAの種類や量を調べるのが目的光刺激 ヒト AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA 転写物全体 ( トランスクリプトーム ) 遺伝子 は光刺激に応答して発現亢進 遺伝子 4 も光刺激に応答して発現亢進 mrna 78
179 トランスクリプトーム解析 光刺激前 (T) の目のトランスクリプトーム 遺伝子 遺伝子 遺伝子 3 遺伝子 4 状態の異なる複数サンプルのデータを取得して解析するのが一般的 サンプル間比較 光刺激後 (T) の目のトランスクリプトーム 遺伝子 遺伝子 遺伝子 3 遺伝子 4 79
180 トランスクリプトーム解析 光刺激前 (T) の目のトランスクリプトーム 遺伝子 遺伝子 遺伝子 3 遺伝子 4 具体的な目的は や の発現変動遺伝子同定など これがいわゆる 遺伝子発現行列 光刺激後 (T) の目のトランスクリプトーム 遺伝子 遺伝子 遺伝子 3 遺伝子 4 80
 なので RNA-seq これがいわゆる")

181 データ取得 光刺激前 (T) の目のトランスクリプトーム 遺伝子 遺伝子 遺伝子 3 遺伝子 4 現在は NGS の利用が主流 NGS を用いた RNA の配列決定 (sequencing) なので RNA-seq これがいわゆる 遺伝子発現行列 光刺激後 (T) の目のトランスクリプトーム 遺伝子 遺伝子 遺伝子 3 遺伝子 4 Togo picture gallery by DBCLS is Licensed under a Creative Commons 表示. 日本 (c) 8
182 RNA-seq 概略 入力 : サンプルの RNA 出力 : 大量塩基配列データ 入力 : 抽出された RNA 断片化 出力 : 塩基配列 NGS で配列決定 アダプター付加 Togo picture gallery by DBCLS is Licensed under a Creative Commons 表示. 日本 (c) 8
183 RNA-seq 概略 入力 : 抽出された RNA NGS の出力は リードと呼ばれる 00 塩基程度の短い配列が延々と続く巨大なファイル 各矢印が つのリードに相当 この段階では まだどのリードがどの転写物由来かは不明 ( なので灰色一色 ) 断片化 出力 : 塩基配列 NGS で配列決定 アダプター付加 Togo picture gallery by DBCLS is Licensed under a Creative Commons 表示. 日本 (c) 83
184 遺伝子 転写物 ある状態のあるサンプル ( 例 : 目 ) のあるゲノムの領域 遺伝子 遺伝子 遺伝子 3 遺伝子 4 遺伝子全体 ( ゲノム ) どの染色体上のどの領域にどの遺伝子があるかは調べる個体 ( 例 : ヒト ) が同じなら不変 ( 目だろうが心臓だろうが ) 赤枠部分の表現は 本当は不正確 昔は実験機器の解像度が事実上遺伝子レベルだった 遺伝子発現解析という表現はその名残り ヒト AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA 転写物全体 ( トランスクリプトーム ) 遺伝子 は沢山転写されている ( 発現している ) 遺伝子 4 はごくわずかしか転写されてない mrna 84
185 遺伝子 転写物 ある状態のあるサンプル ( 例 : 目 ) のあるゲノムの領域 遺伝子 遺伝子 遺伝子 3 遺伝子 4 ある遺伝子領域から転写 (transcription) されている転写物 (transcript) は 種類とは限らない ヒト 85
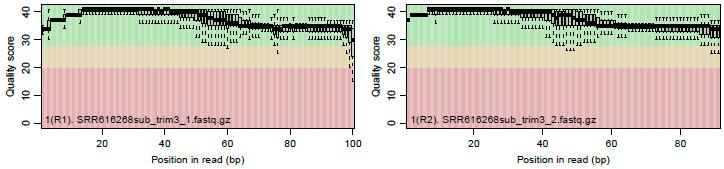
186 遺伝子 転写物 ある状態のあるサンプル ( 例 : 目 ) のあるゲノムの領域 遺伝子 遺伝子 遺伝子 3 遺伝子 4 ある遺伝子領域から転写 (transcription) されている転写物 (transcript) は 種類とは限らない 例えば 遺伝子 の領域では 3 種類の真の転写物が存在し そのうち 種類は既知とする ヒト 遺伝子領域 exon exon exon3 既知転写物 既知転写物 未知転写物 真の転写物情報 86
187 遺伝子 転写物 ある状態のあるサンプル ( 例 : 目 ) のあるゲノムの領域 遺伝子 遺伝子 遺伝子 3 遺伝子 4 実際の細胞内 ( 例 : 目のサンプル ) での発現情報 ( 働いている度合い ) が のような感じだったとする ヒト 遺伝子領域 高発現 exon exon exon3 既知転写物 既知転写物 低発現 未知転写物 中発現 真の転写物情報 真の発現情報 87
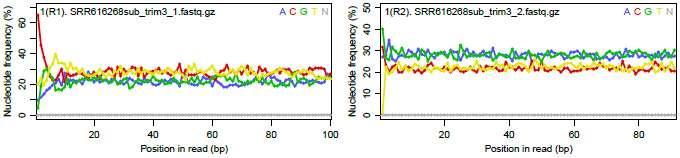
188 遺伝子 転写物 ある状態のあるサンプル ( 例 : 目 ) のあるゲノムの領域 遺伝子 遺伝子 遺伝子 3 遺伝子 4 NGS 機器を用いて転写されている mrna 配列決定 (RNA-seq) をした結果のイメージ ヒト 遺伝子領域 高発現 exon exon exon3 既知転写物 既知転写物 低発現 未知転写物 中発現 真の転写物情報 真の発現情報 RNA-seqで得られるリード情報 ( 色は不明 ) 88
189 データ解析の出発点 トランスクリプトーム (RNA-seq) データ解析の出発点は RNA-seq データファイル RNA-seq データ 89
190 データ解析の出発点 トランスクリプトーム (RNA-seq) データ解析の出発点は RNA-seq データファイル ゲノム配列情報 RNA-seq データ 90
191 データ解析の出発点 遺伝子 遺伝子 遺伝子 3 遺伝子 4 遺伝子領域 トランスクリプトーム (RNA-seq) データ解析の出発点は RNA-seq データファイル ゲノム配列情報 3 ゲノム上のどこにどんな遺伝子 exon 転写物が存在するかというアノテーション情報 exon exon exon3 既知転写物 既知転写物 RNA-seq データ 9
192 解析結果のイメージ RNA-seq データ ゲノム配列情報 3 アノテーション情報を利用して 4 未知転写物 ( 新規 isoform) の同定ができる 遺伝子 遺伝子 遺伝子 3 遺伝子 4 遺伝子領域 exon exon exon3 既知転写物 既知転写物 未知転写物 4 RNA-seq データ 9
193 解析結果のイメージ 遺伝子 遺伝子 遺伝子 3 遺伝子 4 RNA-seq データ ゲノム配列情報 3 アノテーション情報を利用して 4 未知転写物 ( 新規 isoform) の同定ができる 5 転写物の発現量 ( 働いている度合い ) 推定も原理的に可能 遺伝子領域 5 高発現 exon exon exon3 既知転写物 既知転写物 低発現 未知転写物 中発現 RNA-seq データ 93
194 具体的な戦略は? RNA-seq データ ゲノム配列情報 3 アノテーション情報を利用して 4 未知転写物 ( 新規 isoform) の同定ができる 遺伝子 遺伝子 遺伝子 3 遺伝子 4 遺伝子領域 exon exon exon3 既知転写物 既知転写物 未知転写物 4 RNA-seq データ 94
195 具体的な戦略 RNA-seq データ中の 本 本のリード ( 横棒 ) がゲノム上のどの領域から転写されたのかを調べる 文字列検索と本質的に同じであり これがマッピングという作業に相当する ゲノム RNA-seq データ 95
196 具体的な戦略 RNA-seq データ中の 本 本のリード ( 横棒 ) がゲノム上のどの領域から転写されたのかを調べる 文字列検索と本質的に同じであり これがマッピングという作業に相当する ゲノム RNA-seq データ 96
197 具体的な戦略 リードの長さが初期は 35 塩基程度だったが 現在は 50 塩基程度まで伸びている そのおかげで リードを分割してマッピングすることもできる ゲノム RNA-seq データ 97
198 具体的な戦略 分割してマップされたリードは 大抵の場合複数のエクソン (exon) をまたぐリードであり ジャンクションリード (junction read) と呼ばれる ジャンクションリード ゲノム exon exon exon3 RNA-seq データ 98
199 具体的な戦略 既知遺伝子 ( 転写物 ) の座標情報と比較することで 答え合わせも可能 ジャンクションリード ゲノム exon exon exon3 既知転写物 既知転写物 アノテーション情報 ( 既知遺伝子座標情報 ) RNA-seq データ 99
200 具体的な戦略 同様にして 他のジャンクションリードも既知転写物と比較することで ジャンクションリード ゲノム exon exon exon3 既知転写物 既知転写物 アノテーション情報 ( 既知遺伝子座標情報 ) RNA-seq データ 00
201 具体的な戦略 未知転写物 ( 新規 isoform) の同定も原理的に可能 未知転写物?! ジャンクションリード ゲノム exon exon exon3 既知転写物 既知転写物 アノテーション情報 ( 既知遺伝子座標情報 ) RNA-seq データ 0
202 Contents トランスクリプトーム解析 イントロダクション : 簡単な原理 基本イメージ 様々な解析目的 解析データ : 乳酸菌 (L. casei A) QuasRでマッピング ( 基礎 ): コード各部の説明と結果の解釈 QuasRでマッピング ( 応用 ): オプションを指定して実行 カウント情報取得, サンプル間クラスタリング (TCC) 発現変動解析 (TCC) M-A plot モデル 分布 統計的手法 3 群間比較 (TCCによるANOVA 的な解析 ) 遺伝子間クラスタリング (MBCluster.Seq) 3 群間比較 (TCCによるANOVA 的な解析 + MBCluster.Seqでのパターン分類 ) 0
 が動作しており そのさらに内部で基本マッピングプログラムである 3Bowtie( や Bowtie) が動作している 3")
203 様々な解析目的 トランスクリプトーム配列取得 ゲノム配列既知の場合 参考 新規転写物同定などに相当 がメインプログラム 多くのメインプログラム内部で や 3 のサブプログラムが動作する 例えば Bowtie-Tophat-Cufflinks パイプラインは Cufflinks 内部で ジャンクションリードもマップ可能な Tophat( や Tophat) が動作しており そのさらに内部で基本マッピングプログラムである 3Bowtie( や Bowtie) が動作している 3 03
 も出力してくれます")
204 様々な解析目的 トランスクリプトーム配列取得 ゲノム配列未知の場合 参考 トランスクリプトーム配列の de novo アセンブリに相当 多くのプログラムは発現量 (FPKM 値 ) も出力してくれます 04


205 様々な解析目的 発現量の正確な推定 参考 転写物の発現量を正確に推定したい場合は 専用のプログラムを使うべし RSEM が有名 3Sailfish も高速なアルゴリズムとして有名 4TIGER は日本語で質問できる上 最近の手法比較論文 (Kanitz et al. Genome Biol., 05) でも高評価でおススメ

206 様々な解析目的 発現変動解析 ( 群間比較 ) 参考 群間比較で 反復あり ( 複製あり ) データの場合は edger 反復なしの場合は DESeq を内部的に用いて頑健な結果を返す TCC がおススメ 反復の有無に応じて 内部的に用いるパッケージを自動で切り替える 06

207 様々な解析目的 発現変動解析 (3 群間比較 ) 参考 3 群間比較で 反復あり ( 複製あり ) データの場合は edger 反復なしの場合は DESeq を内部的に用いて頑健な結果を返す TCC がおススメ (Tang et al., BMC Bioinformatics, 05) 反復の有無に応じて 内部的に用いるパッケージを自動で切り替える 07
208 解析データ ( 乳酸菌 ) マップする側 (paired-end RNA-seq データ ;SRR6668) オリジナルデータ (Illumina HiSeq 000 で取得 ) の情報 リード長 :forward 側は 07 bp reverse 側は 93 bp リード数 : ともに 34,755,996 リード ( 約.35 億 ) データ量 :bzip 圧縮状態で計約 5GB 非圧縮 FASTQ で計約 80GB 下記手順実行後のデータ ( 計約 0MB) をマッピングに利用. 最初の00 万リードのみ抽出 ( 計 00 万リード ). forward 側 :3 側 7 bpをトリム後にアダプターを除去 998,649リード 3. reverse 側 :3 側 bp をトリム後にアダプターを除去 999,33 リード 4. 両方で存在するリードのみ抽出 998,5 = 計,997,04 リード forward 側 (SRR6668sub_trim3_.fastq.gz) reverse 側 (SRR6668sub_trim3_.fastq.gz) マップされる側 (Lactobacillus casei A) ゲノムサイズ :,907,89 bp 遺伝子数 :,799 個 Lactobacillus casei A 株のデータ 乳酸菌 NGS 連載第 3 回最後のほうでダウンロードしたものと基本的に同じ トリムの理由は第 5 回でわかる Lactobacillus_casei_a.GCA_ dna.chromosome.Chromosome.fa Lactobacillus_casei_a.GCA_ chromosome.Chromosome.gff3 07 bp 93 bp 00 bp 9 bp 08
209 参考 教科書 p-3 FASTA 形式と FASTQ 形式 FASTA 形式がわかるヒトは それに quality 情報のみが追加されたものという理解でよい FASTA 形式 行目 : > ではじまる一行の description 行 行目 : 配列情報 >SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT FASTQ 形式 行目 ではじまる 行の description 行 行目 : 配列情報 3 行目 : + からはじまる 行 ( の description 行 ) 4 行目 : GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT +!''*((((***+))%%%++)(%%%%).***-+*''))**55CCF>>>>>>CCCCCCC

210 解析データ ( 乳酸菌 ) 3 DRAで見たマップする側のpaired-end RNA-seqデータ accession 番号は SRR6668 リード数は に示されている 大元は3SRAという形式で保存されている DRAでは FASTQ 形式ファイルを提供している SRAからFASTQファイルを作成する際に 4でみえているようなNが多くクオリティが低いリードが除去されるため FASTQファイルで取り扱う際にはで見えているリード数よりも若干少なくなる 4 0

211 解析データ ( 乳酸菌 ) マップされる側の ゲノム配列データと アノテーションデータ (GFF ファイル ) 情報は Ensembl から取得 バージョンは年に数回程度以上の頻度で上がっている印象 基本は最新だが 最も重要なのはゲノム配列とアノテーション情報のバージョンが同じであること
212 Contents トランスクリプトーム解析 イントロダクション : 簡単な原理 基本イメージ 様々な解析目的 解析データ : 乳酸菌 (L. casei A) QuasRでマッピング ( 基礎 ): コード各部の説明と結果の解釈 QuasRでマッピング ( 応用 ): オプションを指定して実行 カウント情報取得, サンプル間クラスタリング (TCC) 発現変動解析 (TCC) M-A plot モデル 分布 統計的手法 3 群間比較 (TCCによるANOVA 的な解析 ) 遺伝子間クラスタリング (MBCluster.Seq) 3 群間比較 (TCCによるANOVA 的な解析 + MBCluster.Seqでのパターン分類 )
m 前処理 マッピング QC レポート カウント情報取得まで可能なので")
213 マッピング基礎 QuasR は general なパッケージ 精度云々というよりは Windows の R 環境でマッピングを可能にしてくれたという点で感謝 m( )m 前処理 マッピング QC レポート カウント情報取得まで可能なので このあたりの全貌を大まかに理解するうえで便利 3


214 マッピング基礎 QuasR は basic なマッピングプログラムである bowtie (Langmead et al., Genome Biol., 009) とジャンクションリードをマッピング可能な SpliceMap (Au et al., Nucleic Acids Res., 00) を選択可能 ここではバクテリア ( 乳酸菌 ) ゲノムにマップするので高速な basic aligner を利用 例題 4
215 マッピング基礎 マップされる側のリファレンス配列 マップする側はリストファイル ( タブ区切りテキストファイル ) として与える リストファイルの中身は 3paired-end の場合はこんな感じ 3 5
216 マッピング基礎 リストファイルとして与えるメリットは 複数サンプルの場合を考えればよい のファイルは 行からなるが 3 行目以降に同様な形式で 追加するサンプル数分だけ行を増やしていけばよい SampleName 列は カウントデータ ( 後述 ) を得るときに ここに記載さいたサンプル名 ( ここでは namae_paired) が列名として使われる 6
217 解析データ ( 乳酸菌 ) マップする側 (paired-end RNA-seq データ ;SRR6668) オリジナルデータ (Illumina HiSeq 000 で取得 ) の情報 リード長 :forward 側は 07 bp reverse 側は 93 bp リード数 : ともに 34,755,996 リード ( 約.35 億 ) データ量 :bzip 圧縮状態で計約 5GB 非圧縮 FASTQ で計約 80GB 下記手順実行後のデータ ( 計約 0MB) をマッピングに利用. 最初の00 万リードのみ抽出 ( 計 00 万リード ). forward 側 :3 側 7 bpをトリム後にアダプターを除去 998,649リード 3. reverse 側 :3 側 bp をトリム後にアダプターを除去 999,33 リード 4. 両方で存在するリードのみ抽出 998,5 = 計,997,04 リード forward 側 (SRR6668sub_trim3_.fastq.gz) reverse 側 (SRR6668sub_trim3_.fastq.gz) マップされる側 (Lactobacillus casei A) ゲノムサイズ :,907,89 bp 遺伝子数 :,799 個 マップする側のリストファイルと マップされる側の FASTA 形式ファイル Lactobacillus_casei_a.GCA_ dna.chromosome.Chromosome.fa Lactobacillus_casei_a.GCA_ chromosome.Chromosome.gff3 7


218 マッピング基礎 デスクトップ hoge L.casei_A_genome フォルダ内に必要なファイルはあります 作業ディレクトリをそこに変更して 3list.files() 3 8
 約 4 分")
219 マッピング基礎 赤枠部分が必要最小限 QuasR パッケージを読み込んで 3 主要関数である qalign を実行するだけで BAM 形式のマッピング結果ファイルを得ることができる の赤枠部分をコピペ このプログラムの機能のいくつかが Windows ファイアウォールでブロックされています というアラートウィンドウが出ることもあるが 4 その場合はキャンセルボタンを押す ( でないと先に進めない ) 約 4 分 3 4 9
220 解説 R Console 画面上で見えているのは のあたり マッピング部分の所要時間は 秒 計,997,04 リード中 3693,500 個がマップされ 4,303,54 個がマップされなかったことがわかる 3 4 0
221 解析データ ( 乳酸菌 ) マップする側 (paired-end RNA-seq データ ;SRR6668) オリジナルデータ (Illumina HiSeq 000 で取得 ) の情報 リード長 :forward 側は 07 bp reverse 側は 93 bp リード数 : ともに 34,755,996 リード ( 約.35 億 ) データ量 :bzip 圧縮状態で計約 5GB 非圧縮 FASTQ で計約 80GB 下記手順実行後のデータ ( 計約 0MB) をマッピングに利用. 最初の00 万リードのみ抽出 ( 計 00 万リード ). forward 側 :3 側 7 bpをトリム後にアダプターを除去 998,649リード 3. reverse 側 :3 側 bp をトリム後にアダプターを除去 999,33 リード 4. 両方で存在するリードのみ抽出 998,5 = 計,997,04 リード forward 側 (SRR6668sub_trim3_.fastq.gz) reverse 側 (SRR6668sub_trim3_.fastq.gz) マップされる側 (Lactobacillus casei A) ゲノムサイズ :,907,89 bp 遺伝子数 :,799 個 seqlength の数値は マップされる側のリファレンス配列の総塩基数 Lactobacillus_casei_a.GCA_ dna.chromosome.Chromosome.fa Lactobacillus_casei_a.GCA_ chromosome.Chromosome.gff3
222 解説 qalign 関数実行時に マッピングプログラム (bowtie or SpliceMap) の指定を行わなかった 理由はデフォルトが basic aligner (unspliced aligner ともいう ) の bowtie であることを知っていたから qalign 関数上では splicedalignment = FALSE として表現される 主なマッピング結果である BAM ファイルは 3 拡張子が.bam だが これはバイナリファイルなので中身を眺めても意味不明 ( 爆 ) 3

223 解説 作業フォルダを眺めると 確かに.bam ファイルが作成されている マッピング後の解析は 基本的に BAM 形式ファイルを入力として取り扱う 3 赤下線部分の文字列はヒトそれぞれ 3 3
224 QC レポート PDF 入力データのクオリティスコアやマッピング結果の概要などを PDF ファイルとして出力できます 最後の部分が _QC.pdf となる PDF ファイルが作成されます 4

225 QC レポート PDF 確かに最後の部分が _QC.pdf となる PDF ファイルが作成されていることが分かります これを解説します 5
226 PDF の一部を解説 リードのポジションごとの クオリティスコア分布と 塩基組成 6

227 PDF の一部を解説 マップされたリードの割合 計,997,04 リード (e+06) 中 334.7% がマップされた 4 693,500/,997,04 = なので妥当 3 4 7
 が.3% 43.47e+05 は片側のみで考えているのかもしれない いずれにせよ 5693500 0.")
228 PDF の一部を解説 4 3 マップされたリードのうち か所にのみマップされたリード (uniquely mapped reads) が 87.7% 3 複数個所にマップされたリード (non-unique) が.3% 43.47e+05 は片側のみで考えているのかもしれない いずれにせよ = 個程度はゲノム中の か所にのみマップされたと解釈 5 8


229 BED ファイル作成 BED 形式ファイルは バイナリファイルのため中身を解釈できない BAM ファイルをテキストファイルに変換して可視化したもの という理解でよい 赤枠部分をコピペ.bed というファイルが作成される 3for というループを回しているのは 複数サンプルに対応するため 3 9
")

")
230 BED 概観 作成された BED ファイルをエクセルで眺める マッピング結果は どの配列上の 3 どこから (start) 4 どこまで (end) の場所にリードがマップされたかが最低限わかればよい マップされたリードの総数は 693,500 個だったので ヘッダー行 ( つまり 行目に列名情報 ) がないこのファイルの場合 5693,500 行となるのは妥当
231 Contents トランスクリプトーム解析 イントロダクション : 簡単な原理 基本イメージ 様々な解析目的 解析データ : 乳酸菌 (L. casei A) QuasRでマッピング ( 基礎 ): コード各部の説明と結果の解釈 QuasRでマッピング ( 応用 ): オプションを指定して実行 カウント情報取得, サンプル間クラスタリング (TCC) 発現変動解析 (TCC) M-A plot モデル 分布 統計的手法 3 群間比較 (TCCによるANOVA 的な解析 ) 遺伝子間クラスタリング (MBCluster.Seq) 3 群間比較 (TCCによるANOVA 的な解析 + MBCluster.Seqでのパターン分類 ) 3
 を眺めても -v で指定する許容するミスマッチ数が何だったのか不明 例題 3bowtie (basic aligner の つ ) 利用時に - m -best")
232 マッピング応用 QuasR はオプション無指定でもマッピングを行ってくれるが ログファイル (QuasR_log_be8cf3864.txt) を眺めても -v で指定する許容するミスマッチ数が何だったのか不明 例題 3bowtie (basic aligner の つ ) 利用時に - m -best -strata v 0 オプションをつけて実行する 3 3

233 マッピング応用 以前のマッピング結果が残っている場所で実行してよいが 念のため オブジェクトの全消去をやっておこう 33
 に比べて減っている デフォルトオプション実行時の -v で指定する許容するミスマッチ数は 以上だったのだろうと推測可")
234 マッピング応用 終了後の状態 マップされたリードの総数 (494,9 個 ) は デフォルトの結果 (693,500 個 ) に比べて減っている デフォルトオプション実行時の -v で指定する許容するミスマッチ数は 以上だったのだろうと推測可 34
 中 54.8% がマップされた 494,9/,997,04 = 0.")
235 マッピング応用 赤枠部分が -m -best -strata v 0 オプションをつけて実行した結果ファイル QC レポート PDF 中の 3 マップされたリードの割合 4 計,997,04 リード (e+06) 中 54.8% がマップされた 494,9/,997,04 = なので妥当
236 マッピング応用 マップされたリード ( 片側 47,456 リードで計 494,9 リード ) のうち か所にのみマップされたリード (uniquely mapped reads) が 86.6% 3 複数個所にマップされたリード (non-unique) が 3.4% この結果は直感的にオカシイ 理由は -m として か所にのみマップされるリードを出力しているつもりだから 3 36

237 ? 関数名 Bowtie 実行時のオプションを眺めるべく alignmentparameter のところを詳細に調査 qalign 関数の詳細を調べたいときは 3?qAlign と打つ 数秒後に html マニュアルが開く 3 37
238 関数マニュアル qalign 関数の html マニュアル ページ上部 Usage ( 基本的な利用法 ) Arguments( オプションの説明 ) 3Details( 詳細情報 ) 4Value( 返り値 ; どんな結果が返ってくるか ) などの情報が見られる これはかなり難しい例なので 最初のうちは sum, mean, alphabetfrequency など 挙動を完全に把握できている関数のマニュアルを眺めて慣れておくとよい 38


239 関数マニュアル 調べたい alignmentparameter のところをつぎはぎで表示 結論として -m として か所にのみマップされるリードを出力しているつもりなのに なぜ non-unique が 3.4% 含まれるという結果になるのか理解できない もしかしたら 複数個所にマップされるリードはランダムにどこか か所の結果が返されるということなのだろうか? 少なくとも門田はマニュアルの説明だけでは挙動を完全にイメージできない プログラムのバグかもしれないし 門田の勘違いかもしれないが これ以上は深追いしない 39
240 Contents トランスクリプトーム解析 イントロダクション : 簡単な原理 基本イメージ 様々な解析目的 解析データ : 乳酸菌 (L. casei A) QuasRでマッピング ( 基礎 ): コード各部の説明と結果の解釈 QuasRでマッピング ( 応用 ): オプションを指定して実行 カウント情報取得, サンプル間クラスタリング (TCC) 発現変動解析 (TCC) M-A plot モデル 分布 統計的手法 3 群間比較 (TCCによるANOVA 的な解析 ) 遺伝子間クラスタリング (MBCluster.Seq) 3 群間比較 (TCCによるANOVA 的な解析 + MBCluster.Seqでのパターン分類 ) 40

241 カウント情報取得 アノテーション情報を利用する場合 UCSC known Genes, Ensembl Genes など様々なテーブル名を指定可能 gene, exon, promoter, junction など様々なレベルを指定可能 アノテーション情報がない場合 R でのアノテーション情報利用は TxDb が基本 マップされたリードの和集合領域を同定したのち 領域ごとのリード数をカウント BEDtools (Quinlan et al., 00) 中の mergebed プログラムを実行して和集合領域同定後 intersectbed プログラムを実行してリード数をカウントする作業に相当 count 領域 3 4 4

242 カウント情報取得 アノテーション情報を利用する場合 UCSC known Genes, Ensembl Genes など様々なテーブル名を指定可能 gene, exon, promoter, junction など様々なレベルを指定可能 アノテーション情報がない場合 アノテーション情報がない場合の戦略は 複数サンプルの場合には領域が変わりうる Cufflinks を知っているヒトは cuffmerge と同じイメージだと思えばよい マップされたリードの和集合領域を同定したのち 領域ごとのリード数をカウント BEDtools (Quinlan et al., 00) 中の mergebed プログラムを実行して和集合領域同定後 intersectbed プログラムを実行してリード数をカウントする作業に相当 sample count sample 4
243 カウント情報取得 アノテーション情報を利用するやり方の 例題 をやってみましょう 43
244 違いを説明 マッピング基礎 の例題 と基本的に同じ 違いは GFF 形式のアノテーションファイルの指定 カウントデータ取得のレベルを指定 3 アノテーションファイルの読み込み 3 44
 で指定したレベルのカウントデータを取得する qcount 実行部分 および 結果の保存の部分")
245 違いを説明 マッピング結果 out とアノテーション情報 txdb を読み込んで param_reportlevel( この場合 gene レベル ) で指定したレベルのカウントデータを取得する qcount 実行部分 および 結果の保存の部分 45

246 コピペで実行 同じデフォルトオプションで実行した マッピング基礎 の例題 の結果がある 作業ディレクトリ上で 念のためオブジェクトの全消去を行ってからコピペ 分弱 46

247 無駄を省く 分弱で終わる理由は マッピングを行っていないから QuasR はまずログファイルを調べる そして以前に同じ入力ファイル 同じオプションで実行した結果を見つけたら (QuasR_log_be8cf3864.txt) ( あなたが実行したい ) 全てのマッピング結果が見つかったよ となり 以前の結果を読み込んでくれる 47

248 無駄を省く マッピング基礎 の例題 の結果と同じく 693,500 リードがマップされた out オブジェクトを利用可能なので 3qCount 関数でカウントデータを得ることができる 3 48
249 qcount qcount 関数実行によって得られたカウントデータ情報を含む count オブジェクトの 行数と列数は 77 行 列 3count オブジェクトの最初の 6 行分を head 関数で表示 3 49
250 qcount 列目は配列長 列目が目的のカウント情報 3param_reportlevel で指定していたのは 4gene それゆえ 5 行名は 遺伝子名の通し番号のようになっているのだろう
251 qcount 遺伝子名の通し番号っぽいものは txdb オブジェクトの元情報である 3in_f3 で読み込んだアノテーションファイル中に書かれている筈 3 5
252 GFF を眺めて確認 GFF ファイルをエクセルで眺めている の文字列が out_f で指定した出力ファイル (hoge.txt) 中の 3 行名として使われているのだろう 3 5

253 GFF を眺めて確認 基本テクニックを駆使して 配列長 350 bpの LCAA_067を 確認 同じ配列長になっており妥当 完全に欠番のない通し番号になっていれば3count 行列の67 行目という指定でもイケる わけではない 4tailで納得
254 Contents トランスクリプトーム解析 イントロダクション : 簡単な原理 基本イメージ 様々な解析目的 解析データ : 乳酸菌 (L. casei A) QuasRでマッピング ( 基礎 ): コード各部の説明と結果の解釈 QuasRでマッピング ( 応用 ): オプションを指定して実行 カウント情報取得, サンプル間クラスタリング (TCC) 発現変動解析 (TCC) M-A plot モデル 分布 統計的手法 3 群間比較 (TCCによるANOVA 的な解析 ) 遺伝子間クラスタリング (MBCluster.Seq) 3 群間比較 (TCCによるANOVA 的な解析 + MBCluster.Seqでのパターン分類 ) 54

255 カウント情報取得 さきほどまでは例題 の解説 次は 例題 4 55
256 カウント情報取得 例題 4 は マッピング応用 の例題 と同じくマッピング時のオプションを明示的に指定している 許容するミスマッチ数を 0(-v 0) にしたときのマップされたリード数 ( 494,9 個 ) は デフォルトの結果 (693,500 個 ) よりも少なかった カウント行列データの数値は全体的に少なくなると予想できるので それを確認するのが主目的 56
257 カウント情報取得 コピペで実行し 全体像を見られるところを表示 マップされたリード数は 確かに 494,9 個だった 3 カウント行列データの数値は確かに全体的に少ない 3 57

258 比較 許容するミスマッチ数を 0(-v 0) にしたときの結果のほうが 確かに デフォルトの結果に比べて少ない 58
259 出力ファイル 例題 と 例題 4 のもう つの違いは 配列長 (hoge4_length.txt) と 3 カウント情報 (hoge4_count.txt) を別々に出力している点 3 59
260 出力ファイル カウント情報ファイル (hoge4_count.txt) の中身はこんな感じ 配列情報の列を含まないものが一般的なカウントデータ以降の統計解析の入力ファイルとして利用される 60
261 Contents トランスクリプトーム解析 イントロダクション : 簡単な原理 基本イメージ 様々な解析目的 解析データ : 乳酸菌 (L. casei A) QuasRでマッピング ( 基礎 ): コード各部の説明と結果の解釈 QuasRでマッピング ( 応用 ): オプションを指定して実行 カウント情報取得, サンプル間クラスタリング (TCC) 発現変動解析 (TCC) M-A plot モデル 分布 統計的手法 3 群間比較 (TCCによるANOVA 的な解析 ) 遺伝子間クラスタリング (MBCluster.Seq) 3 群間比較 (TCCによるANOVA 的な解析 + MBCluster.Seqでのパターン分類 ) 6
 は 3 種類の生物種間比較 0,689 genes 8samples ヒト (HS) チンパンジー (PT) アカゲザル (RM) 生物種ごとにメス 3 匹 オス 3 匹 発現変動遺伝子")
262 カウントデータ解析 カウントデータ このデータ (Blekhman et al., 00) は 3 種類の生物種間比較 0,689 genes 8samples ヒト (HS) チンパンジー (PT) アカゲザル (RM) 生物種ごとにメス 3 匹 オス 3 匹 発現変動遺伝子 (DEG) 同定 サンプル間クラスタリング Blekhman et al., Genome Res., 0: 80-89, 00 6


 のサンプル間クラスタリング")
263 サンプル間クラスタリング 0,689 遺伝子 8 サンプルの biological replicates のみからなるカウントデータ (Blekhman et al., 00) のサンプル間クラスタリング データの取得や整形については の講義資料を参考 63

264 入力ファイル デスクトップ上の hoge フォルダ中に sample_blekhman_8.txt が存在するはず ファイルが存在しないヒトは 右クリック 3 対象をファイルに保存 でデスクトップ上の hoge に保存 3 Blekhman et al., Genome Res., 0: 80-89, 00 64
265 0,689 genes 入力ファイル このデータは 3 種類の生物種間比較 ヒト (Homo sapiens; HS) チンパンジー (Pan troglodytes; PT) アカゲザル (Rhesus macaque; RM) 生物種ごとにメス 3 匹 オス 3 匹 雄雌を考慮しなければ biological replicates ( 生物学的な反復 ) は 6 ヒト (Homo sapiens; HS) チンパンジー (Pan troglodytes; PT) アカゲザル (Rhesus macaque; RM) メス (Female) オス (Male) メスオスメスオス Blekhman et al., Genome Res., 0: 80-89, 00 65


266 コピペで実行 一連のコマンド群をコピーして R Console 画面上でペースト ブラウザが Internet Explorer の場合は CTRL と ALT キーを押しながらコードの枠内で左クリックすると 全選択できます 66

267 実行結果 エラーなく実行できると右下のような画面になっているはずです 入力ファイル情報を格納した行列 data の行数が 0,689 列数が 8 となっていることがわかります 67
268 400 ピクセル 出力ファイル 出力ファイルのサイズを指定しているので こんな感じになる hoge8.png 700 ピクセル 68
269 結果の解釈 ヒト (HS) オス3 匹 (M, M, M3) メス3 匹 (F, F, F3) チンパンジー (PT) オス3 匹 (M, M, M3) メス3 匹 (F, F, F3) アカゲザル (RM) オス3 匹 (M, M, M3) メス3 匹 (F, F, F3) hoge8.png 3 生物種間全体で眺めると ヒト (HS) とチンパンジー (PT) はよく似ている ヒト (HS) チンパンジー (PT) アカゲザル (RM) 69
270 結果の解釈 ヒト (HS) オス3 匹 (M, M, M3) メス3 匹 (F, F, F3) チンパンジー (PT) オス3 匹 (M, M, M3) メス3 匹 (F, F, F3) アカゲザル (RM) オス3 匹 (M, M, M3) メス3 匹 (F, F, F3) hoge8.png 群間比較 ( 発現変動遺伝子検出 ; DEG 検出 ) を行ったときに HS vs. RM で得られる DEG 数 のほうが 3 HS vs. PT で得られる DEG 数 よりも多そう ヒト (HS) チンパンジー (PT) アカゲザル (RM) 3 70
271 結果の解釈 ヒト (HS) オス3 匹 (M, M, M3) メス3 匹 (F, F, F3) チンパンジー (PT) オス3 匹 (M, M, M3) メス3 匹 (F, F, F3) アカゲザル (RM) オス3 匹 (M, M, M3) メス3 匹 (F, F, F3) hoge8.png 同一生物種でクラスターを形成している RMM は 外れサンプル っぽい ヒト (HS) チンパンジー (PT) アカゲザル (RM) 7
272 結果の解釈 ヒト (HS) オス3 匹 (M, M, M3) メス3 匹 (F, F, F3) チンパンジー (PT) オス3 匹 (M, M, M3) メス3 匹 (F, F, F3) アカゲザル (RM) オス3 匹 (M, M, M3) メス3 匹 (F, F, F3) hoge8.png ヒト (HS) と アカゲザル (RM) は メスとオスのサンプルが入り混じっている これらの生物種内で メス群 vs. オス群 の 群間比較を行っても DEG はほとんど検出されないだろう ヒト (HS) チンパンジー (PT) アカゲザル (RM) 7
273 結果の解釈 ヒト (HS) オス3 匹 (M, M, M3) メス3 匹 (F, F, F3) チンパンジー (PT) オス3 匹 (M, M, M3) メス3 匹 (F, F, F3) アカゲザル (RM) オス3 匹 (M, M, M3) メス3 匹 (F, F, F3) hoge8.png チンパンジー (PT) に限っていえば メス 3 匹がクラスターを形成しているので メス群 vs. オス群 の 群間比較結果として 多少なりとも DEG が検出されるだろう ヒト (HS) チンパンジー (PT) アカゲザル (RM) 73
274 この解釈の仕方は 原著論文あり 但し TCC パッケージが提供する clustersample 関数を用いた結果以外では責任を持ちません また 運悪く例外データセットもあるかも Tang et al., BMC Bioinformatics, 6: 36, 05 74
 の Additional file 6 に 全く同じ図もあり つまり コピペで作成したクラスタリング結果がそのまま論文の図に使えるということ 3")
275 論文レベルの図です 手法比較原著論文 (Tang et al., 05) の Additional file 6 に 全く同じ図もあり つまり コピペで作成したクラスタリング結果がそのまま論文の図に使えるということ 3 書き方は赤下線のような感じでよい 3 Tang et al., BMC Bioinformatics, 6: 36, 05 75
276 Contents トランスクリプトーム解析 イントロダクション : 簡単な原理 基本イメージ 様々な解析目的 解析データ : 乳酸菌 (L. casei A) QuasRでマッピング ( 基礎 ): コード各部の説明と結果の解釈 QuasRでマッピング ( 応用 ): オプションを指定して実行 カウント情報取得, サンプル間クラスタリング (TCC) 発現変動解析 (TCC) M-A plot モデル 分布 統計的手法 3 群間比較 (TCCによるANOVA 的な解析 ) 遺伝子間クラスタリング (MBCluster.Seq) 3 群間比較 (TCCによるANOVA 的な解析 + MBCluster.Seqでのパターン分類 ) 76

277 HS vs. RM HS vs. RM の 群間比較を TCC パッケージ (Sun et al., 03) で行ってみよう 77
278 サブセット抽出 例題 黒枠内をコピペ 78
279 サブセット抽出 ここで取得したいサブセットの列番号やグループ情報を指定 発現変動解析に用いるサブセットは 0,689 genes 4 samples のデータ 3 正しくヒト (HS) vs. アカゲザル (RM) になっている 3 79
 を眺めるなどして")
280 サブセット抽出 入力ファイル (sample_blekhman_8.txt) を眺めるなどして 該当サンプルの列の位置を把握していることが前提 80
 = 0.")
 とみなすと,488*0.05 = 4.")
281 FDR 赤枠部分までをコピペ q < 0.05 を満たす遺伝子数は,488 個 False discovery rate (FDR) = 0.05 は この閾値を満たす,488 個を発現変動遺伝子 (Differentially Expressed Genes; DEGs) とみなすと,488*0.05 = 4.4 個は偽物であることを意味する 有意水準 (false positive rate; FPR)5% と同じような位置づけであり FDR5% というのは 許容する偽物 (non-deg) 混入割合 に相当する 8

282 FDR 最後までコピペ q < 0.30 を満たす遺伝子数は 4,786 個 FDR = 0.30 なので 4,786*0.30 =,435.8 個は偽物で残りの 70% は本物だと判断する 8
283 DEG 数の見積もり FDR 閾値が比較的緩めのところを眺め 0,689 genes 中 3,300 個程度が本物の DEG と判断する 83
284 樹形図と一致 今比較しているのは HS vs. RM クラスタリング結果からも これらの発現プロファイルの類似度が低い ( 距離が遠い ) ので妥当 84
285 Contents トランスクリプトーム解析 イントロダクション : 簡単な原理 基本イメージ 様々な解析目的 解析データ : 乳酸菌 (L. casei A) QuasRでマッピング ( 基礎 ): コード各部の説明と結果の解釈 QuasRでマッピング ( 応用 ): オプションを指定して実行 カウント情報取得, サンプル間クラスタリング (TCC) 発現変動解析 (TCC) M-A plot モデル 分布 統計的手法 3 群間比較 (TCCによるANOVA 的な解析 ) 遺伝子間クラスタリング (MBCluster.Seq) 3 群間比較 (TCCによるANOVA 的な解析 + MBCluster.Seqでのパターン分類 ) 85
286 M-A plot これが M-A plot 発現変動遺伝子 (DEG) と判定されたものが多数存在することがわかる param_fdr で指定した閾値 (0.05) を満たす遺伝子群がマゼンタ色で表示されている 86
 横軸が平均 (Average) G 群 < G 群 G 群で高発現 G 群 = G 群 G 群 > G 群 G 群で高発現 3 4 5 A = (log G + log G)/ 低発現 全体的に 高発現 Dudoit et al., Stat.")
287 M = log G - log G M-A plot DEG が存在しないデータの M-A plot を眺めることで 縦軸の閾値のみに相当する倍率変化を用いた DEG 同定の危険性が分かります 群間比較用 横軸が全体的な発現レベル 縦軸が log 比からなるプロット 名前の由来は おそらく対数の世界での縦軸が引き算 (Minus) 横軸が平均 (Average) G 群 < G 群 G 群で高発現 G 群 = G 群 G 群 > G 群 G 群で高発現 A = (log G + log G)/ 低発現 全体的に 高発現 Dudoit et al., Stat. Sinica, : -39, 00 87
288 DEG 検出結果 基本的にはこれが解析結果 88

289 DEG 検出結果 G(HS) 群 G(RM) 群 位は RM 群 (G 群 ) で高発現の DEG p-value とその順位 G 群で高発現 M-A plot の A 値と M 値 q-value FDR 閾値判定結果 q-value < 0.05 を満たす DEG が non-deg が 0 G 群で高発現 89
290 DEG 検出結果 G(HS) 群 G(RM) 群 位も RM 群 (G 群 ) で高発現の DEG p-value とその順位 G 群で高発現 M-A plot の A 値と M 値 q-value FDR 閾値判定結果 q-value < 0.05 を満たす DEG が non-deg が 0 G 群で高発現 90
291 DEG 検出結果 G(HS) 群 G(RM) 群 3 位は HS 群 (G 群 ) で高発現の DEG p-value とその順位 G 群で高発現 M-A plot の A 値と M 値 q-value FDR 閾値判定結果 q-value < 0.05 を満たす DEG が non-deg が 0 G 群で高発現 9
 をギリギリ満たす,488 位の遺伝子 p-value とその順位 G 群で高発現 M-A plot の A 値と M 値 q-value FDR 閾値判定結果")
292 DEG 検出結果 G(HS) 群 G(RM) 群 指定した FDR 閾値 (0.05) をギリギリ満たす,488 位の遺伝子 p-value とその順位 G 群で高発現 M-A plot の A 値と M 値 q-value FDR 閾値判定結果 q-value < 0.05 を満たす DEG が non-deg が 0 G 群で高発現 9

293 様々な M-A plot 例題 -5 のコピペで作成しました 93
294 Contents トランスクリプトーム解析 イントロダクション : 簡単な原理 基本イメージ 様々な解析目的 解析データ : 乳酸菌 (L. casei A) QuasRでマッピング ( 基礎 ): コード各部の説明と結果の解釈 QuasRでマッピング ( 応用 ): オプションを指定して実行 カウント情報取得, サンプル間クラスタリング (TCC) 発現変動解析 (TCC) M-A plot モデル 分布 統計的手法 3 群間比較 (TCCによるANOVA 的な解析 ) 遺伝子間クラスタリング (MBCluster.Seq) 3 群間比較 (TCCによるANOVA 的な解析 + MBCluster.Seqでのパターン分類 ) 94
295 分布やモデル ( 当たり前だが )FDR 閾値を緩めると得られる DEG 数は増える傾向にあることがわかる 例題 6 のコピペで作成 HS vs. RM HS vs. RM HS vs. RM 73 DEGs (FDR 0.0%),488 DEGs (FDR 5%) 5,435 DEGs (FDR 40%) 厳しい FDR 閾値 緩い 少ない DEG 数 多い 95
,488 DEGs (FDR 5%) 5,435 DEGs (FDR 40%) 厳しい FDR 閾値 緩い 少ない DEG 数 多い 96")
296 分布やモデル 重要 : 黒の分布は non-deg の分布に相当 HS vs. RM HS vs. RM HS vs. RM 73 DEGs (FDR 0.0%),488 DEGs (FDR 5%) 5,435 DEGs (FDR 40%) 厳しい FDR 閾値 緩い 少ない DEG 数 多い 96
297 統計的手法とは HS vs. RM HS vs. PT 同一群 ( 下段 ) の分布は 異なる群 ( 上段 ) の non-deg 分布とよく一致する 同一群内のばらつきの分布 (non-deg 分布 ) 以外のものが DEG と判定されるのが統計的手法の結果,488 DEGs 578 DEGs ヒト (HS) チンパンジー (PT) HS vs. HS PT vs. PT RM vs. RM RM vs. RM3 アカゲザル (RM) 7 DEGs 6 DEGs 4 DEGs 0 DEGs 97
298 統計的手法とは 例題 3 同一群内のばらつきの分布 (non- DEG 分布 ) から遠く離れたところに位置するものは 0 に近い p-value 同一群内の遺伝子のばらつきの程度を把握し 帰無仮説に従う分布の全体像を把握しておく ( モデル構築 ) non-deg のばらつきの程度を把握しておくことと同義 実際に比較したい 群の遺伝子のばらつきの程度が non-deg 分布のどのあたりに位置するかを評価 ( 検定 ) HS vs. RM 98
 non-deg のばらつきの程度を把握しておくことと同義 実際に比較したい 群の遺伝子のばらつきの程度が non-deg")
299 統計的手法とは 例題 同一群内のばらつきの分布 (non- DEG 分布 ) の ど真ん中に位置するものは に近い p-value 同一群内の遺伝子のばらつきの程度を把握し 帰無仮説に従う分布の全体像を把握しておく ( モデル構築 ) non-deg のばらつきの程度を把握しておくことと同義 実際に比較したい 群の遺伝子のばらつきの程度が non-deg 分布のどのあたりに位置するかを評価 ( 検定 ) 99
 チンパンジー (PT) HS vs. HS PT vs. PT RM vs. RM RM vs.")
300 結果の比較 ( 倍変化 ) 倍率変化 (fold-change; FC) での DEG 検出結果 下段の同一群内比較でも多数の偽陽性が検出されている 例題 3 をベースに作成 HS vs. RM HS vs. PT 5,966 DEGs 5,077 DEGs ヒト (HS) チンパンジー (PT) HS vs. HS PT vs. PT RM vs. RM RM vs. RM3 アカゲザル (RM),7 DEGs 3,375 DEGs 3,806 DEGs 3,00 DEGs 300
301 結果の比較 (FDR) HS vs. RM HS vs. PT 統計的手法 (TCC) も多少偽陽性が存在するが 倍率変化 (FC) ほど凶悪ではないことがわかる また高発現側の DEG は FC と比較的よく一致していることがわかる 先人が FC のみで比較的信頼性の高い結果を得てきた理由がよくわかる ( 高発現側を信頼するという経験則 ),488 DEGs 578 DEGs ヒト (HS) チンパンジー (PT) HS vs. HS PT vs. PT RM vs. RM RM vs. RM3 アカゲザル (RM) 7 DEGs 6 DEGs 4 DEGs 0 DEGs 30
302 Contents トランスクリプトーム解析 イントロダクション : 簡単な原理 基本イメージ 様々な解析目的 解析データ : 乳酸菌 (L. casei A) QuasRでマッピング ( 基礎 ): コード各部の説明と結果の解釈 QuasRでマッピング ( 応用 ): オプションを指定して実行 カウント情報取得, サンプル間クラスタリング (TCC) 発現変動解析 (TCC) M-A plot モデル 分布 統計的手法 3 群間比較 (TCCによるANOVA 的な解析 ) 遺伝子間クラスタリング (MBCluster.Seq) 3 群間比較 (TCCによるANOVA 的な解析 + MBCluster.Seqでのパターン分類 ) 30
303 0,689 genes 3 群間比較 このデータは 3 種類の生物種間比較 : ヒト (Homo sapiens; HS) チンパンジー (Pan troglodytes; PT) アカゲザル (Rhesus macaque; RM) どこかの群間で発現変動している遺伝子を検出するやり方を示す ヒト (Homo sapiens; HS) チンパンジー (Pan troglodytes; PT) アカゲザル (Rhesus macaque; RM) メス (Female) オス (Male) メスオスメスオス Blekhman et al., Genome Res., 0: 80-89,
304 3 群間比較論文 3 群間比較用に特化した手法選択のガイドライン 反復ありデータの場合は ( 内部的に edger の関数を用いた )TCC 反復なしの場合は ( 内部的に DESeq を用いた )TCC がおススメ Tang et al., BMC Bioinformatics, 6: 36,
305 データ正規化周辺 RPM (Mortazavi et al., Nat. Methods, 5: 6-68, 008) RPKM(Reads per kilobase of exon per million mapped reads) の長さ補正を行わないバージョン Reads Per Million mapped reads の略 TMM 正規化 (Robinson and Oshlack, Genome Biol., : R5, 00) Trimmed Mean of M values の略 edger パッケージに実装されている 発現変動遺伝子 (DEG) のデータ正規化時の悪影響を排除すべく M-A plot 上で周縁部にあるデータを使わずに ( トリムして ) 正規化係数を決定する方法 TbT 正規化 (Kadota et al., Algorithms Mol. Biol., 7: 5, 0) TMM 法の改良版で TMM-baySeq-TMM という 3 ステップで正規化を行う方法 st step で得られた TMM 正規化係数を用いて nd step (bayseq) で DEG 同定を行い 3rd step (TMM) では DEG を排除した残りのデータで TMM 正規化 DEG の影響を排除しつつもできるだけ多くの non- DEG データを用いて頑健に正規化係数を決めるという思想 (DEG elimination strategy 提唱論文 ) ideges 正規化 (Sun et al., BMC Bioinformatics, 4: 9, 03) TCC パッケージの原著論文 参考 発現変動解析 ( サンプル間比較 ) 時に重要となる sequence depth 周辺の正規化法の進展 の HPCI セミナースライドから拝借 DEG elimination strategy (DEGES) を一般化し より高速且つ頑健にしたもの TbT は 複製あり のデータのみにしか対応していなかったが 複製なし データにも対応 ideges/edger 正規化法 : 複製あり データ正規化用 TMM-(edgeR-TMM) n パイプライン ideges/deseq 正規化法 : 複製なし データ正規化用 DESeq-(DESeq-DESeq) n パイプライン 305
306 TbT 正規化法 参考 TCC パッケージに実装している基本コンセプトの原著論文 TbT 正規化法の説明 のイルミナウェビナー時のスライドの拝借 本来の目的である発現変動遺伝子 (DEG) 自体がデータ正規化時に悪影響を与えるので DEG 候補を除去して正規化を行うほうがよいこと (DEG Elimination Strategy) を提唱した論文 既存の正規化法は 比較するグループ間で DEG 数に偏りがない (unbiased DE) 場合にはうまく正規化できるが 偏りがある場合 (biased DE) にはうまく正規化できないことを示した TbT 法の実体は edger パッケージ中の TMM 正規化法実行 bayseq パッケージ中の DEG 検出法実行 および 3DEG 候補を除去した残りの non-deg 候補のみを用いた TMM 正規化法実行 の 3 ステップを基本とする TMM-baySeq-TMM パイプライン 出力は正規化後の結果 ( 正確には正規化係数 ) なので TbT 正規化後に任意の DEG 検出法を適用することで一連の発現変動解析が終了することになる 例えば TbT 正規化法実行後に edger 中の DEG 検出法を適用する一連の手順は TMM-baySeq-TMM-edgeR に相当し 原著論文中では edger/tbt と略記している 論文中では TbT にした理由を論理的に書いたが 本音は ToT に近いものということで TMM と bayseq を採用 提案したマルチステップの正規化パイプラインは 第 および第 3 ステップを繰り返して実行することでより頑健な正規化を実現可能であることも示している これが図 3 で説明している iterative TbT approach に相当するものであり TMM-(baySeq-TMM)n とも表現できる 例えば iterative TbT 正規化法実行後に edger 中の DEG 検出法を適用する一連の手順は TMM-(baySeq- TMM)n-edgeR に相当する n = 0 の場合は TMM-edgeR となり これは edger パッケージ中のオリジナルの手順と同じである 306
307 TCC 参考 TbT 論文の考え方を一般化し R パッケージとしてまとめたという論文 TbT 正規化法の説明 のイルミナウェビナー時のスライドの拝借 TbT は DEG Elimination Strategy (DEGES; でげす ) に基づく一つの正規化パイプラインにすぎないこと 第 ステップの bayseq による DEG 同定ステップが律速であり高速化が課題であったこと そして各ステップにおいて他の方法が原理的に適用可能であることなどを述べている 第 ステップの DEG 同定法を edger 中のものに置き換えると TMM-edgeR-TMM という正規化パイプラインになる これは 全て edger パッケージ中の関数のみで成立するため DEGES/edgeR と略記している また DEGES 正規化後に edger 中の DEG 同定法を適用する一連の解析手順は DEGES/edgeR-edgeR または TMM-edgeR-TMM-edgeR と表記できる これは実質的に edger パッケージ中のオリジナルの解析手順を 回繰り返して行っていることと同義である ( ただし 第 3 ステップの TMM は検出された DEG 候補以外のデータで実行される ) それが 実質的に TCC は例えば iterative edger という理解でよい と主張する根拠である TbT 論文中で言及した iterative TbT に相当するものは この論文中では iterative DEGES ( 略して ideges) と称している 例えば ideges/edger-edger は TMM-(edgeR-TMM)n-edgeR に相当する n= は DEGES/edgeR-edgeR に相当する n が 以上の場合が ideges に相当するが n の数を増やしてもその分計算コストがかかる一方で 実質的に n=3 程度で頭打ちになることを論文中で示している それゆえ iterative DEGES のデフォルトは n=3 としている compcoder (Soneson, C., Bioinformatics, 04) 中でもデフォルトはそうなっている 307
308 0,689 genes 3 群間比較 sample_blekhman_8.txt を入力として ヒト (HS)6 サンプル チンパンジー (PT)6 サンプル アカゲザル (RM)6 サンプルの 3 群間比較を行います どこかの群間で発現変動している遺伝子を検出するやり方です 各群のサンプルは全て別個体です 例えばヒトの場合は 6 人分のデータ (6 biological replicates) であり 人のサンプルを 6 個に分割したデータ (6 technical replicates) ではありません ヒト (Homo sapiens; HS) チンパンジー (Pan troglodytes; PT) アカゲザル (Rhesus macaque; RM) Blekhman et al., Genome Res., 0: 80-89,



309 3 群間比較 例題 7 3 入力は sample_blekhman_8.txt 出力は hoge7.txt のみ M-A plot はない 3 Sun et al., BMC Bioinformatics, 4: 9, 03 Tang et al., BMC Bioinformatics, 6: 36,
310 3 群間比較 ここで各群のサンプル数 ( 列数 ) を指定 FDR 閾値を指定するところだが 出力ファイルの読み取り方が分かっていれば気にしなくてもよい Sun et al., BMC Bioinformatics, 4: 9, 03 Tang et al., BMC Bioinformatics, 6: 36, 05 30
311 3 群間比較 コピペで実行した結果の最後の部分を表示 約 分 赤枠部分は 4 種類の FDR 閾値を満たす遺伝子数を表示している 例えば は 5% の偽物混入を許容すると 7,47 個が DEG と判定されるということ 3

312 DEG 数の見積もり FDR 閾値が比較的緩めのところを眺め 0,689 genes 中 8,000 個程度がどこかの群間で発現変動している本物の DEG と判断する 3
313 出力ファイル解説 出力ファイル (hoge7.txt) の中身を解説 正規化後のデータ 3 統計解析結果 (p 値 q 値 順位情報など ) 4 発現変動順にソートされた結果
314 コードの解説 正規化後のデータを取り出す部分の説明 データ正規化 ( 正確には正規化係数を得た ) 後の tcc オブジェクトを入力として getnormalizeddata 関数を用いて正規化後のデータを取得した結果を normalized オブジェクトに格納し 3 出力の一部として組み込んでいる
315 コードの解説 ( このウェブページではお約束的にそうしているので ) 番左側に行名をしているが 実は の result オブジェクトの 3 番左側が同じ情報なのでなくてもよい 3 35
316 コードの解説 rank( 順位 ) 情報は p 値をベースに計算している p 値が低いほど発現変動の度合いが高いことを意味する 実際 3 位の発現パターンはチンパンジー (PT) 群で 4 桁以上のカウント数で それ以外の群では 桁となっており妥当 3 36
317 ANOVA 的な解析 位はアカゲザル (RM) 群で高発現のパターン 3 位はヒト (HS) 群で高発現のパターン ( どの群間で違いがあるかは問わずに ) どこかの群間で発現に差があるものを検出する枠組み (ANOVA 的な解析 ) なので このような結果になる 37
 結果を tmp オブジェクトに格納しているだけ 実際 result")
318 コードの解説 の段階では まだ発現変動順にはソートされておらず cbind 関数を用いて列方向で結合した (column bind) 結果を tmp オブジェクトに格納しているだけ 実際 result オブジェクトは入力ファイルの遺伝子名の並びになっている 38
319 コードの解説 ( 赤字のコメント部分をよく見ればわかるが w) 発現変動順にソートしている部分がココ tmp[order(x), ] は x の並びで行をソートするお約束的な書き方 発現変動順にソートしたくない場合は 行頭に # をつけてコメントアウトすればよい 39
320 コードの解説 の部分が灰色になっているのは あくまでも 0.05, 0.0, 0.0, 0.30 という FDR 閾値を満たす遺伝子数をざっと表示させて概要 (DEG があるかどうか あったとしたらどれくらいか ) を知りたいだけだから 30
321 コードの解説 の部分は 出力ファイル中の q.value 列を見ているのと同じ 3
322 コードの解説 例えば 5% FDR threshold というのは q-value (q 値 ) が 0.05 未満 (or 以下 ) という条件判定を行っていることと同義 これは有意水準 5% というのが 実際の手順として p-value(p 値 ) < 0.05 という条件判定を行っていることを思い出せば納得できる 3
323 コードの解説 番右側の estimateddeg という名前の列が q-value (q 値 ) < 0.05 のところで から 0 に切り替わっているのは param_fdr のところで 0.05 を指定していたから の指定部分を気にしなくてよいと最初に解説したのは q-value 列の取り扱い方法を理解していればそれで充分だからです 33
324 パターン分類 ( どの群間で違いがあるかは問わずに ) どこかの群間で発現に差があるものを検出する枠組み (ANOVA 的な解析 ) なのでこのような結果になる のはしょうがいないとして G(HS) 群で高発現のものが x 個 G(PT) 群で高発現のものが y 個 みたいなものが欲しい! 34
325 Contents トランスクリプトーム解析 イントロダクション : 簡単な原理 基本イメージ 様々な解析目的 解析データ : 乳酸菌 (L. casei A) QuasRでマッピング ( 基礎 ): コード各部の説明と結果の解釈 QuasRでマッピング ( 応用 ): オプションを指定して実行 カウント情報取得, サンプル間クラスタリング (TCC) 発現変動解析 (TCC) M-A plot モデル 分布 統計的手法 3 群間比較 (TCCによるANOVA 的な解析 ) 遺伝子間クラスタリング (MBCluster.Seq) 3 群間比較 (TCCによるANOVA 的な解析 + MBCluster.Seqでのパターン分類 ) 35
326 遺伝子クラスタリング ( 個人的には結論として非推奨だが ) 遺伝子間クラスタリングを行っておき どの遺伝子がどのパターンに属するかという情報を MBCluster.Seq などを用いて得ておき 特定の FDR 閾値を満たす遺伝子サブセットの分類分けを行えばよい おそらくこれも王道なので一応紹介 36


327 遺伝子クラスタリング MBCluster.Seq 単体での利用はこちら 例題 4 3K-means クラスタリングの一種なので 3 クラスター数を指定する 50 など比較的大きめの値にしても non-redundant にしてくれるので 最終的に得られるクラスター数は ( データの性質にもよるが通常は ) 減る 3 37
328 遺伝子クラスタリング 通常のクラスタリングの際は 入力データのどの列がどの群かという情報を与えない MBCluster.Seq は 同一群内のバラツキを考慮してくれるので 列のラベル情報を与えている Model-based clustering というのは ( 発現変動解析時に non-deg の分布を見積もるのと同様に ) 同一群内のバラツキを超えた意味のある発現パターンを見積もって返してくれると理解すればよい 38
 コピペ 約")
329 コピペ (CTRL + ALT + 左クリック またはトリプルクリックでコードを全選択して ) コピペ 約 8 分 39
330 結果の説明 ピクセルの hoge4.png 内部的に乱数を発生させているので 見栄えはヒトによって異なる 330
331 結果の説明 下から順に G(HS) G(PT) G3(RM) 順序は のテキストファイル ( 後述 ) と見比べることでわかる 3cluster と 0 は G3 群で高発現 4cluster 6 と 8 は G3 群で低発現パターンのものだ みたいに解釈する 全体を眺めることで各クラスタを構成するメンバー数 ( 遺伝子数 ) の概要をつかめる 例えば 5cluster 9 を構成する遺伝子数が最多 とか
332 3 結果の説明 テキストファイル (hoge4.txt) の中身 番右側の列がクラスター番号情報 3 例題 4 の場合 出力ファイルはクラスター番号順にソートされている 確かにデンドログラム ( 樹形図 ) でみた通り cluster は G3(RM) 群で高発現パターンになっている 33
 作成部分のコード data オブジェクトは入力ファイルを読み込んだ直後のものなので 正規化前のデータ 3cls$cluster という数値ベクトルが 入力データの遺伝子の並び順に")
333 コードの解説 テキストファイル (hoge4.txt) 作成部分のコード data オブジェクトは入力ファイルを読み込んだ直後のものなので 正規化前のデータ 3cls$cluster という数値ベクトルが 入力データの遺伝子の並び順に どの遺伝子がどのクラスターに属するかを示した情報に相当する 出力がクラスター番号順になっている理由は 4cls$cluster の並びでソートしているから 門田亡き後もこのようにコードの中身を自力で解読できるようになっておけば大丈夫
334 クラスタごとの遺伝子数 hoge4.png を眺めることで cluster 9 を構成する遺伝子数が最多とかが一応わかるが 各クラスタを構成するメンバー数 ( 遺伝子数 ) を正確に調べるやり方を伝授 334
 であることからもこの結果が妥当であることが分かります この結果も乱数を発生させているので ヒトによって結果は異なる 3 は理解を助ける補足情報 3")
335 クラスタごとの遺伝子数 入力データの遺伝子の並び順にどの遺伝子がどのクラスターに属するかを示した cls$cluster という数値ベクトルを入力として table 関数を実行した結果が欲しいものです を眺めると 確かに cluster 9 のメンバー数が最多 (7,67 個 ) であることからもこの結果が妥当であることが分かります この結果も乱数を発生させているので ヒトによって結果は異なる 3 は理解を助ける補足情報 3 335
336 Contents トランスクリプトーム解析 イントロダクション : 簡単な原理 基本イメージ 様々な解析目的 解析データ : 乳酸菌 (L. casei A) QuasRでマッピング ( 基礎 ): コード各部の説明と結果の解釈 QuasRでマッピング ( 応用 ): オプションを指定して実行 カウント情報取得, サンプル間クラスタリング (TCC) 発現変動解析 (TCC) M-A plot モデル 分布 統計的手法 3 群間比較 (TCCによるANOVA 的な解析 ) 遺伝子間クラスタリング (MBCluster.Seq) 3 群間比較 (TCCによるANOVA 的な解析 + MBCluster.Seqでのパターン分類 ) 336



337 TCC + MBCluster.Seq 応用 の項目の 例題 8 が TCC でどこかの群間で発現変動する遺伝子と MBCluster.Seq を組み合わせたものです コピペ 約 0 分 337

338 TCC + MBCluster.Seq ピクセルの hoge8.png 内部的に乱数を発生させているので 見栄えはヒトによって異なる この結果の場合は 3cluster 7 が non-deg パターンであり 最多の遺伝子数から構成される 3 338
 の中身 TCC の結果部分は不変")
339 TCC + MBCluster.Seq テキストファイル (hoge8.txt) の中身 TCC の結果部分は不変 一番右側の列の数値はおそらくヒトによって異なる 339
340 クラスタごとの遺伝子数 コードの最後の部分を表示 全遺伝子 (0,689 個 ) を対象としたクラスターごとの遺伝子数 png ファイルで眺めた non- DEG パターンに相当する cluster 7 の遺伝子数が最多 (7,43 個 ) となっており妥当 340
341 クラスタごとの遺伝子数 5% FDR 閾値を満たす遺伝子 (7,47 個 ) に限定して クラスターごとの遺伝子数を再分類 non-deg パターンに相当する cluster 7 の遺伝子数が最少 (67 個 ) となっており妥当 34
(H7-9 年度 ): ロングリード時代に対応したトランスクリプトームデータ解析ガイドラインの構築 ( 代表 ) 基盤研究 (C)(H4-6 年度 ): シークエンスに基づく比較トランスクリプトーム解析のためのガイドライン構築 ( 代表 ) 新学術領域研究 ( 研究領域提案型 )(H-6 年度 ):")
342 共同研究者 謝辞 所属 敬称略 他にもバグレポートやプログラム提供をいただいた諸氏 R 本体および有用なパッケージ開発者諸氏に御礼申し上げます m( )m アグリバイオ本体 TCC パッケージ および手法比較清水謙多郎 寺田透 三浦文 孫建強 西山智明 湯敏 DDBJ Pipeline Platanus および日本乳酸菌学会誌谷澤靖洋 神沼英里 中村保一 遠野雅徳 有田正規 伊藤武彦 鈴木チセ 坂本光央 HPCI 人材養成プログラム杉原稔 寺田朋子グラント 基盤研究 (C)(H7-9 年度 ): ロングリード時代に対応したトランスクリプトームデータ解析ガイドラインの構築 ( 代表 ) 基盤研究 (C)(H4-6 年度 ): シークエンスに基づく比較トランスクリプトーム解析のためのガイドライン構築 ( 代表 ) 新学術領域研究 ( 研究領域提案型 )(H-6 年度 ): 非モデル生物におけるゲノム解析法の確立 ( 分担 ; 研究代表者 : 西山智明 ) NBDC との共同研究 (H6-7 年度 ):NGS 講習会関連 34
基本的な利用法
 (R で ) 塩基配列解析 基本的な利用法 Macintosh 版 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける 1. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール 2. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握
(R で ) 塩基配列解析 基本的な利用法 Macintosh 版 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける 1. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール 2. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握
基本的な利用法
 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握 ) 4. ありがちなミス のところで示しているエラーメッセージとその原因をきっちり理解
到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握 ) 4. ありがちなミス のところで示しているエラーメッセージとその原因をきっちり理解
Rインストール手順
 R 本体は最新のリリース版 R パッケージは 必要最小限プラスアルファ の推奨インストール手順を示します R 本体とパッケージのインストール Windows 版 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/
R 本体は最新のリリース版 R パッケージは 必要最小限プラスアルファ の推奨インストール手順を示します R 本体とパッケージのインストール Windows 版 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/
ゲノム情報解析基礎 ~ Rで塩基配列解析 ~
 ネット接続できないヒトも ダブルクリックでローカルに r_seq.html を起動可能です 実習は デスクトップ上にある hoge フォルダの中身が以下の状態を想定して行います (R で ) 塩基配列解析の利用法 : GC 含量計算から発現変動解析まで東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
ネット接続できないヒトも ダブルクリックでローカルに r_seq.html を起動可能です 実習は デスクトップ上にある hoge フォルダの中身が以下の状態を想定して行います (R で ) 塩基配列解析の利用法 : GC 含量計算から発現変動解析まで東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
機能ゲノム学
 08.05.08 版 講義資料 PDF が講義のページからダウンロード可能です 講義資料の印刷物はありません 課題用の A4 一枚はあります 第 回出席予定の持込み PC の方は 当日までに Java のインストールをしておいてください 機能ゲノム学第 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム 微生物科学イノベーション連携研究機構門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
08.05.08 版 講義資料 PDF が講義のページからダウンロード可能です 講義資料の印刷物はありません 課題用の A4 一枚はあります 第 回出席予定の持込み PC の方は 当日までに Java のインストールをしておいてください 機能ゲノム学第 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム 微生物科学イノベーション連携研究機構門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
ゲノム情報解析基礎
 講義資料 PDF が講義のページからダウンロード可能です 印刷物はありません ゲノム情報解析基礎 ~ R で塩基配列解析 ~ 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム 微生物科学イノベーション連携研究機構門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 講義予定
講義資料 PDF が講義のページからダウンロード可能です 印刷物はありません ゲノム情報解析基礎 ~ R で塩基配列解析 ~ 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム 微生物科学イノベーション連携研究機構門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 講義予定
基本的な利用法
 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握 ) 4. ありがちなミス のところで示しているエラーメッセージとその原因をきっちり理解
到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握 ) 4. ありがちなミス のところで示しているエラーメッセージとその原因をきっちり理解
NGS速習コース
 バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-3. R 各種パッケージ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-3. R 各種パッケージ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
農学生命情報科学特論I
 2015.07.01 版 USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 前回 (6/23) の hoge フォルダがデスクトップに残っているかもしれないのでご注意ください 農学生命情報科学 特論 I 第 3 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
2015.07.01 版 USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 前回 (6/23) の hoge フォルダがデスクトップに残っているかもしれないのでご注意ください 農学生命情報科学 特論 I 第 3 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
NGS速習コース
 バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-4. R Bioconductor I 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-4. R Bioconductor I 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
NGSハンズオン講習会
 207.08.08 版 プラスアルファの内容です NGS 解析 ( 初 ~ 中級 ) ゲノムアセンブリ後の各種解析の補足資料 ( プラスアルファ ) 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム寺田朋子 門田幸二 Aug 29-30 207 Contents Gepard でドットプロット 連載第 8 回 W5-3 で最も長い sequence 同士のドットプロットを実行できなかったが
207.08.08 版 プラスアルファの内容です NGS 解析 ( 初 ~ 中級 ) ゲノムアセンブリ後の各種解析の補足資料 ( プラスアルファ ) 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム寺田朋子 門田幸二 Aug 29-30 207 Contents Gepard でドットプロット 連載第 8 回 W5-3 で最も長い sequence 同士のドットプロットを実行できなかったが
NGSデータ解析入門Webセミナー
 NGS データ解析入門 Web セミナー : RNA-Seq 解析編 1 RNA-Seq データ解析の手順 遺伝子発現量測定 シークエンス マッピング サンプル間比較 機能解析など 2 CLC Genomics Workbench 使用ツール シークエンスデータ メタデータのインポート NGS data import Import Metadata クオリティチェック Create Sequencing
NGS データ解析入門 Web セミナー : RNA-Seq 解析編 1 RNA-Seq データ解析の手順 遺伝子発現量測定 シークエンス マッピング サンプル間比較 機能解析など 2 CLC Genomics Workbench 使用ツール シークエンスデータ メタデータのインポート NGS data import Import Metadata クオリティチェック Create Sequencing
NGSハンズオン講習会
 205.07.27 版 配布する USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください NGS ハンズオン 講習会 :R 基礎 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ Contents(
205.07.27 版 配布する USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください NGS ハンズオン 講習会 :R 基礎 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ Contents(
特論I
 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 2 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG 概論 現状や展望など 講義のみ 第 2 回 (2014
講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 2 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG 概論 現状や展望など 講義のみ 第 2 回 (2014
機能ゲノム学(第6回)
") トランスクリプトーム解析の今昔 なぜマイクロアレイ? なぜRNA-Seq? 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 Contents トランスクリプトーム解析の概要 各手法の長所 短所 マイクロアレイ
トランスクリプトーム解析の今昔 なぜマイクロアレイ? なぜRNA-Seq? 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 Contents トランスクリプトーム解析の概要 各手法の長所 短所 マイクロアレイ
特論I
 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 4 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp 1 前回の課題と正答 アダプター配列除去前後の small RNA-seq データをカイコゲノムにマップし マップ率 ( マップされたリード数
講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 4 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp 1 前回の課題と正答 アダプター配列除去前後の small RNA-seq データをカイコゲノムにマップし マップ率 ( マップされたリード数
Rインストール手順
 R 本体は最新のリリース版 R パッケージは 必要最小限プラスアルファ の推奨インストール手順を示します スライドは Windows0 環境でのスクリーンショットです ウェブブラウザによって挙動が多少異なるのでご注意ください 私は Chrome を使っています R 本体とパッケージのインストール Windows 版 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二
R 本体は最新のリリース版 R パッケージは 必要最小限プラスアルファ の推奨インストール手順を示します スライドは Windows0 環境でのスクリーンショットです ウェブブラウザによって挙動が多少異なるのでご注意ください 私は Chrome を使っています R 本体とパッケージのインストール Windows 版 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二
シーケンサー利用技術講習会 第10回 サンプルQC、RNAseqライブラリー作製/データ解析実習講習会
 シーケンサー利用技術講習会 第 10 回サンプル QC RNAseq ライブ ラリー作製 / データ解析実習講習会 理化学研究所ライフサイエンス技術基盤研究センターゲノムネットワーク解析支援施設田上道平 次世代シーケンサー Sequencer File Format Output(Max) Read length Illumina Hiseq2500 Fastq 600 Gb 100 bp Life
シーケンサー利用技術講習会 第 10 回サンプル QC RNAseq ライブ ラリー作製 / データ解析実習講習会 理化学研究所ライフサイエンス技術基盤研究センターゲノムネットワーク解析支援施設田上道平 次世代シーケンサー Sequencer File Format Output(Max) Read length Illumina Hiseq2500 Fastq 600 Gb 100 bp Life
Rでゲノム・トランスクリプトーム解析
 R でゲノム トランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 自己紹介 1995 年 3 月 高知工業高等専門学校 工業化学科卒業 1997 年 3 月 東京農工大学 工学部 物質生物工学科卒業
R でゲノム トランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 自己紹介 1995 年 3 月 高知工業高等専門学校 工業化学科卒業 1997 年 3 月 東京農工大学 工学部 物質生物工学科卒業
1. はじめに 1. はじめに 1-1. KaPPA-Average とは KaPPA-Average は KaPPA-View( でマイクロアレイデータを解析する際に便利なデータ変換ソフトウェアです 一般のマイクロアレイでは 一つのプロー
 KaPPA-Average 1.0 マニュアル 第 1.0 版 制作者 : かずさ DNA 研究所櫻井望 制作日 : 2010 年 1 月 12 日 目次 1. はじめに 2 1-1. KaPPA-Average とは 2 1-2. 動作環境 3 1-3. インストールと起動 3 2. 操作説明 4 2-1. メイン機能 - Calc. Average 4 2-1-1. データの準備 4 2-1-2.
KaPPA-Average 1.0 マニュアル 第 1.0 版 制作者 : かずさ DNA 研究所櫻井望 制作日 : 2010 年 1 月 12 日 目次 1. はじめに 2 1-1. KaPPA-Average とは 2 1-2. 動作環境 3 1-3. インストールと起動 3 2. 操作説明 4 2-1. メイン機能 - Calc. Average 4 2-1-1. データの準備 4 2-1-2.
ゲノム情報解析基礎
 ゲノム情報解析基礎 ~ R で塩基配列解析 ~ 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 多くのヒトが感想を述べられて 感想やコメント へのコメントいました ありがとうございます コピペではなく位置から自分が入力するのは無理そう
ゲノム情報解析基礎 ~ R で塩基配列解析 ~ 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 多くのヒトが感想を述べられて 感想やコメント へのコメントいました ありがとうございます コピペではなく位置から自分が入力するのは無理そう
特論I
 2016.02.01 版 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 3 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp Jun 25, 2014 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG
2016.02.01 版 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 3 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp Jun 25, 2014 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG
機能ゲノム学(第6回)
") RNA-Seq データ解析リテラシー 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 2009 年ごろの私 次世代シーケンサー (NGS) 解析についての認識 単に短い塩基配列が沢山あるだけでしょ 得られる配列データって
RNA-Seq データ解析リテラシー 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 2009 年ごろの私 次世代シーケンサー (NGS) 解析についての認識 単に短い塩基配列が沢山あるだけでしょ 得られる配列データって
Rでゲノム・トランスクリプトーム解析
 06.07.06 版 第 3 部 :NGS 解析 ( 中 ~ 上級 ) ~ クラウド環境との連携 ロングリードデータの解析 ~ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ Aug 03 06, NGS
06.07.06 版 第 3 部 :NGS 解析 ( 中 ~ 上級 ) ~ クラウド環境との連携 ロングリードデータの解析 ~ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ Aug 03 06, NGS
機能ゲノム学(第6回)
") R でトランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 自己紹介 2002 年 3 月 東京大学 大学院農学生命科学研究科博士課程修了 学位論文 : cdna マイクロアレイを用いた遺伝子発現解析手法の開発
R でトランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 自己紹介 2002 年 3 月 東京大学 大学院農学生命科学研究科博士課程修了 学位論文 : cdna マイクロアレイを用いた遺伝子発現解析手法の開発
GWB
 NGS データ解析入門 Web セミナー : De Novo シークエンス解析編 1 NGS 新規ゲノム配列解析の手順 シークエンス 遺伝子領域の検出 アセンブル データベース検索 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 前処理 コンティグ配列の作成 CLC Genomics Workbench 遺伝子領域の検出 Blast2GO PRO データベース検索
NGS データ解析入門 Web セミナー : De Novo シークエンス解析編 1 NGS 新規ゲノム配列解析の手順 シークエンス 遺伝子領域の検出 アセンブル データベース検索 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 前処理 コンティグ配列の作成 CLC Genomics Workbench 遺伝子領域の検出 Blast2GO PRO データベース検索
PowerPoint プレゼンテーション
 V1 次世代シークエンサ実習 II 本講義の内容 Reseq 解析 RNA-seq 解析 公開データ取得 クオリティコントロール マッピング 変異検出 公開データ取得 クオリティコントロール マッピング 発現定量 FPKM を算出します 2 R N A - s e q とは メッセンジャー RNA(mRNA) をキャプチャして次世代シーケンサーでシーケンシングする手法 リファレンスがある生物種の場合
V1 次世代シークエンサ実習 II 本講義の内容 Reseq 解析 RNA-seq 解析 公開データ取得 クオリティコントロール マッピング 変異検出 公開データ取得 クオリティコントロール マッピング 発現定量 FPKM を算出します 2 R N A - s e q とは メッセンジャー RNA(mRNA) をキャプチャして次世代シーケンサーでシーケンシングする手法 リファレンスがある生物種の場合
IonTorrent RNA-Seq 解析概要 サーモフィッシャーサイエンティフィックライフテクノロジーズジャパンテクニカルサポート The world leader in serving science
 IonTorrent RNA-Seq 解析概要 2017-03 サーモフィッシャーサイエンティフィックライフテクノロジーズジャパンテクニカルサポート The world leader in serving science 資料概要 この資料は IonTorrent シーケンサーで RNA-Seq (WholeTranscriptome mrna ampliseqrna mirna) 解析を実施されるユーザー様向けの内容となっています
IonTorrent RNA-Seq 解析概要 2017-03 サーモフィッシャーサイエンティフィックライフテクノロジーズジャパンテクニカルサポート The world leader in serving science 資料概要 この資料は IonTorrent シーケンサーで RNA-Seq (WholeTranscriptome mrna ampliseqrna mirna) 解析を実施されるユーザー様向けの内容となっています
win版8日目
 8 日目 : 項目のチェック (2) 1 日 30 分くらい,30 日で何とか R をそこそこ使えるようになるための練習帳 :Win 版 昨日は, 平均値などの基礎統計量を計算する試行錯誤へご招待しましたが (?), 今日は簡 単にやってみます そのためには,psych というパッケージが必要となりますが, パッケー ジのインストール & 読み込みの詳しい方法は, 後で説明します 以下の説明は,psych
8 日目 : 項目のチェック (2) 1 日 30 分くらい,30 日で何とか R をそこそこ使えるようになるための練習帳 :Win 版 昨日は, 平均値などの基礎統計量を計算する試行錯誤へご招待しましたが (?), 今日は簡 単にやってみます そのためには,psych というパッケージが必要となりますが, パッケー ジのインストール & 読み込みの詳しい方法は, 後で説明します 以下の説明は,psych
GWB
 NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 CLC Genomics Workbench 使用ツール シークエンスデータのインポート NGS data import クオリティチェック QC for Sequencing Reads Trim Reads 参照ゲノム配列へのマッピング 再アライメント
NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 CLC Genomics Workbench 使用ツール シークエンスデータのインポート NGS data import クオリティチェック QC for Sequencing Reads Trim Reads 参照ゲノム配列へのマッピング 再アライメント
PrimerArray® Analysis Tool Ver.2.2
 研究用 PrimerArray Analysis Tool Ver.2.2 説明書 v201801 PrimerArray Analysis Tool Ver.2.2 は PrimerArray( 製品コード PH001 ~ PH007 PH009 ~ PH015 PN001 ~ PN015) で得られたデータを解析するためのツールで コントロールサンプルと 1 種類の未知サンプル間の比較が可能です
研究用 PrimerArray Analysis Tool Ver.2.2 説明書 v201801 PrimerArray Analysis Tool Ver.2.2 は PrimerArray( 製品コード PH001 ~ PH007 PH009 ~ PH015 PN001 ~ PN015) で得られたデータを解析するためのツールで コントロールサンプルと 1 種類の未知サンプル間の比較が可能です
GWB_RNA-Seq_
 CLC Genomics Workbench ウェブトレーニングセミナー : RNA-Seq 編 フィルジェン株式会社バイオサイエンス部 (biosupport@filgen.jp) 1 Advanced RNA-Seq プラグイン CLC Genomics Workbench 9.0 / Biomedical Genomics Workbench 3.0 以降で使用可能な無償プラグイン RNA-Seq
CLC Genomics Workbench ウェブトレーニングセミナー : RNA-Seq 編 フィルジェン株式会社バイオサイエンス部 (biosupport@filgen.jp) 1 Advanced RNA-Seq プラグイン CLC Genomics Workbench 9.0 / Biomedical Genomics Workbench 3.0 以降で使用可能な無償プラグイン RNA-Seq
リード・ゲノム・アノテーションインポート
 リード ゲノム アノテーションインポート 1 Location と Folder ロケーション フォルダ Genomics Workbenchではデータを以下のような階層構造で保存可能です フォルダの一番上位の階層を Location と呼び その下の階層を Folder と呼びます データの保存場所はロケーション毎に設定可能です たとえばあるデータは C ドライブに保存し あるデータは D ドライブに保存するといった事が可能です
リード ゲノム アノテーションインポート 1 Location と Folder ロケーション フォルダ Genomics Workbenchではデータを以下のような階層構造で保存可能です フォルダの一番上位の階層を Location と呼び その下の階層を Folder と呼びます データの保存場所はロケーション毎に設定可能です たとえばあるデータは C ドライブに保存し あるデータは D ドライブに保存するといった事が可能です
機能ゲノム学(第6回)
") RNA-Seqデータ解析における正規化法の選択 :RPKM 値でサンプル間比較は危険?! 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 よりよい正規化法とは? その正規化法によって得られたデータを用いて発現変動の度合いでランキングしたときに
RNA-Seqデータ解析における正規化法の選択 :RPKM 値でサンプル間比較は危険?! 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 よりよい正規化法とは? その正規化法によって得られたデータを用いて発現変動の度合いでランキングしたときに
バイオインフォマティクス(学部)
") 06.09.0 版 バイオインフォマティクス ~Linux で NGS 解析 ( の基礎 )~ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ Contents イントロダクション 概要 背景 (NGS 用カリキュラム
06.09.0 版 バイオインフォマティクス ~Linux で NGS 解析 ( の基礎 )~ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ Contents イントロダクション 概要 背景 (NGS 用カリキュラム
ChIP-seq
 ChIP-seq 1 ChIP-seq 解析原理 ChIP サンプルのフラグメントでは タンパク質結合部位付近にそれぞれ Forward と Reverse のリードがマップされることが予想される ChIP のサンプルでは Forward と Reverse のリードを 3 側へシフトさせ ChIP のピークを算出する コントロールサンプルでは ChIP のサンプルとは異なり 特定の場所に多くマップされないため
ChIP-seq 1 ChIP-seq 解析原理 ChIP サンプルのフラグメントでは タンパク質結合部位付近にそれぞれ Forward と Reverse のリードがマップされることが予想される ChIP のサンプルでは Forward と Reverse のリードを 3 側へシフトさせ ChIP のピークを算出する コントロールサンプルでは ChIP のサンプルとは異なり 特定の場所に多くマップされないため
_unix_text_command.pptx
 Unix によるテキストファイル処理 2015/07/30 作業場所 以降の作業は 以下のディレクトリで行います ~/unix15/text/ cd コマンドを用いてディレクトリを移動し pwd コマンドを利用して カレントディレクトリが上記になっていることを確認してください 実習で使用するデータ 講習で使用するデータは以下のフォルダ内 ファイルがあることを確認してください ~/unix15/text/
Unix によるテキストファイル処理 2015/07/30 作業場所 以降の作業は 以下のディレクトリで行います ~/unix15/text/ cd コマンドを用いてディレクトリを移動し pwd コマンドを利用して カレントディレクトリが上記になっていることを確認してください 実習で使用するデータ 講習で使用するデータは以下のフォルダ内 ファイルがあることを確認してください ~/unix15/text/
インストールマニュアル
 Install manual by SparxSystems Japan Enterprise Architect 日本語版インストールマニュアル 1 1. はじめに このインストールマニュアルは Enterprise Architect 日本語版バージョン 14.1 をインストールするための マニュアルです インストールには管理者権限が必要です 管理者権限を持つユーザー (Administrator
Install manual by SparxSystems Japan Enterprise Architect 日本語版インストールマニュアル 1 1. はじめに このインストールマニュアルは Enterprise Architect 日本語版バージョン 14.1 をインストールするための マニュアルです インストールには管理者権限が必要です 管理者権限を持つユーザー (Administrator
第21章 表計算
 第 3 部 第 3 章 Web サイトの作成 3.3.1 WEB ページ作成ソフト Dreamweaver の基本操作 Web ページは HTML CSS という言語で作成されており これらは一般的なテキストエディタで作成できるのが特徴ですが その入 力 編集は時に煩雑なものです そこで それらの入力 編集作業など Web ページの作成を補助するソフトウェアである Dreamweaver の使い方について解説していきます
第 3 部 第 3 章 Web サイトの作成 3.3.1 WEB ページ作成ソフト Dreamweaver の基本操作 Web ページは HTML CSS という言語で作成されており これらは一般的なテキストエディタで作成できるのが特徴ですが その入 力 編集は時に煩雑なものです そこで それらの入力 編集作業など Web ページの作成を補助するソフトウェアである Dreamweaver の使い方について解説していきます
引き算アフィリ ASP 登録用の日記サイトを 作成しよう Copyright 株式会社アリウープ, All Rights Reserved. 1
 引き算アフィリ ASP 登録用の日記サイトを 作成しよう 1 目次 ASP 登録用の日記サイトを作成しよう... 3 日記サイト作成時のポイント... 4 (1) 子ページを5ページ分作成する... 5 (2)1 記事当たり600 文字以上書く... 6 (3) アップロードする場所はドメインのトップが理想... 7 日記サイトを作成しよう... 8 日記サイト用テンプレートをダウンロードする...
引き算アフィリ ASP 登録用の日記サイトを 作成しよう 1 目次 ASP 登録用の日記サイトを作成しよう... 3 日記サイト作成時のポイント... 4 (1) 子ページを5ページ分作成する... 5 (2)1 記事当たり600 文字以上書く... 6 (3) アップロードする場所はドメインのトップが理想... 7 日記サイトを作成しよう... 8 日記サイト用テンプレートをダウンロードする...
内容 1 はじめに インストールの手順 起動の手順 Enterprise Architect のプロジェクトファイルを開く 内容を参照する プロジェクトブラウザを利用する ダイアグラムを開く 便利な機能.
 Viewer manual by SparxSystems Japan Enterprise Architect 読み込み専用版 (Viewer) 利用マニュアル 内容 1 はじめに...3 2 インストールの手順...3 3 起動の手順...6 4 Enterprise Architect のプロジェクトファイルを開く...7 5 内容を参照する...8 5.1 プロジェクトブラウザを利用する...8
Viewer manual by SparxSystems Japan Enterprise Architect 読み込み専用版 (Viewer) 利用マニュアル 内容 1 はじめに...3 2 インストールの手順...3 3 起動の手順...6 4 Enterprise Architect のプロジェクトファイルを開く...7 5 内容を参照する...8 5.1 プロジェクトブラウザを利用する...8
Microsoft Word - CygwinでPython.docx
 Cygwin でプログラミング 2018/4/9 千葉 数値計算は計算プログラムを書いて行うわけですが プログラムには様々な 言語 があるので そのうちどれかを選択する必要があります プログラム言語には 人間が書いたプログラムを一度計算機用に翻訳したのち計算を実行するものと 人間が書いたプログラムを計算機が読んでそのまま実行するものとがあります ( 若干不正確な説明ですが ) 前者を システム言語
Cygwin でプログラミング 2018/4/9 千葉 数値計算は計算プログラムを書いて行うわけですが プログラムには様々な 言語 があるので そのうちどれかを選択する必要があります プログラム言語には 人間が書いたプログラムを一度計算機用に翻訳したのち計算を実行するものと 人間が書いたプログラムを計算機が読んでそのまま実行するものとがあります ( 若干不正確な説明ですが ) 前者を システム言語
Outlook2010 の メール 連絡先 に関連する内容を解説します 注意 :Outlook2007 と Outlook2010 では 基本操作 基本画面が違うため この資料では Outlook2010 のみで参考にしてください Outlook2010 の画面構成について... 2 メールについて
 Outlook2010 - メール 連絡先など - Outlook2010 の メール 連絡先 に関連する内容を解説します 注意 :Outlook2007 と Outlook2010 では 基本操作 基本画面が違うため この資料では Outlook2010 のみで参考にしてください Outlook2010 の画面構成について... 2 メールについて... 3 画面構成と操作... 3 人物情報ウィンドウ...
Outlook2010 - メール 連絡先など - Outlook2010 の メール 連絡先 に関連する内容を解説します 注意 :Outlook2007 と Outlook2010 では 基本操作 基本画面が違うため この資料では Outlook2010 のみで参考にしてください Outlook2010 の画面構成について... 2 メールについて... 3 画面構成と操作... 3 人物情報ウィンドウ...
データ科学2.pptx
 データ科学 多重検定 2 mul%ple test False Discovery Rate 藤博幸 前回の復習 1 多くの検定を繰り返す時には 単純に個々の検定を繰り返すだけでは不十分 5% 有意水準ということは, 1000 回検定を繰り返すと, 50 回くらいは帰無仮説が正しいのに 間違って棄却されてすまうじちがあるということ ex) 1 万個の遺伝子について 正常細胞とガン細胞で それぞれの遺伝子の発現に差があるかどうかを検定
データ科学 多重検定 2 mul%ple test False Discovery Rate 藤博幸 前回の復習 1 多くの検定を繰り返す時には 単純に個々の検定を繰り返すだけでは不十分 5% 有意水準ということは, 1000 回検定を繰り返すと, 50 回くらいは帰無仮説が正しいのに 間違って棄却されてすまうじちがあるということ ex) 1 万個の遺伝子について 正常細胞とガン細胞で それぞれの遺伝子の発現に差があるかどうかを検定
数量的アプローチ 年 6 月 11 日 イントロダクション データ分析をマスターする 12 のレッスン ウェブサポートページ ( 有斐閣 ) 水落研究室 R http:
 水落研究室 R http:") イントロダクション データ分析をマスターする 12 のレッスン ウェブサポートページ ( 有斐閣 ) http://yuhikaku-nibu.txt-nifty.com/blog/2017/09/22103.html 水落研究室 R http://depts.nanzan-u.ac.jp/ugrad/ps/mizuochi/r.html 1 この授業では統計ソフト R を使って分析を行います データを扱うソフトとして
イントロダクション データ分析をマスターする 12 のレッスン ウェブサポートページ ( 有斐閣 ) http://yuhikaku-nibu.txt-nifty.com/blog/2017/09/22103.html 水落研究室 R http://depts.nanzan-u.ac.jp/ugrad/ps/mizuochi/r.html 1 この授業では統計ソフト R を使って分析を行います データを扱うソフトとして
プログラマブル LED 制御モジュール アプリ操作説明書 プログラマブル LED 制御モジュール設定アプリ操作説明書 適用モジュール 改訂番号 エレラボドットコム 1
 設定 適用モジュール 041-1 改訂番号 20161024 エレラボドットコム 1 ( 用アプリの利用可能環境 ) Windows7 8.1 10 のいずれかが動作する PC Windows8 以降の場合は 次ページ記載の Windows8 以降の.NET Framework の有効化 (p3~7) の操作をするか 設定されていることを確認してからアプリをインストールしてください.NET Framework2.0
設定 適用モジュール 041-1 改訂番号 20161024 エレラボドットコム 1 ( 用アプリの利用可能環境 ) Windows7 8.1 10 のいずれかが動作する PC Windows8 以降の場合は 次ページ記載の Windows8 以降の.NET Framework の有効化 (p3~7) の操作をするか 設定されていることを確認してからアプリをインストールしてください.NET Framework2.0
RNA-seq
 RNA-seq 1 RNA-seq 解析フロー RNA-seq インポート クオリティチェック RNA-seq 発現差解析 この資料では RNA-seq からの説明となりますが インポート クオリティチェックについては サポート資料のページより内容をご確認いただけます 2 データ 発現解析用デモデータは 以下よりダウンロードいただけます ES 細胞 (ESC) と神経前駆細胞 (NPC) の発現解析を小さなデモデータで行えます
RNA-seq 1 RNA-seq 解析フロー RNA-seq インポート クオリティチェック RNA-seq 発現差解析 この資料では RNA-seq からの説明となりますが インポート クオリティチェックについては サポート資料のページより内容をご確認いただけます 2 データ 発現解析用デモデータは 以下よりダウンロードいただけます ES 細胞 (ESC) と神経前駆細胞 (NPC) の発現解析を小さなデモデータで行えます
第 2 回 (4/18) 実力診断の解説と表作成の復習
 実力診断の解説と表作成の復習") 第 2 回 (4/18) 実力診断の解説と表作成の復習 Gmail の設定を見直す Gmail の必要と思われる設定 送信元情報 署名 Gmail での設定変更 画面右上の歯車マークをクリック 設定 送信元情報と署名 メール設定 アカウント 名前 メール設定 全般 署名 最低限 氏名とアドレスは書こう スレッド表示の無効化 ( 任意 ) スレッド表示とは 関連性のあるメールを合わせて表示 実際にはうまくいかないことが多い
第 2 回 (4/18) 実力診断の解説と表作成の復習 Gmail の設定を見直す Gmail の必要と思われる設定 送信元情報 署名 Gmail での設定変更 画面右上の歯車マークをクリック 設定 送信元情報と署名 メール設定 アカウント 名前 メール設定 全般 署名 最低限 氏名とアドレスは書こう スレッド表示の無効化 ( 任意 ) スレッド表示とは 関連性のあるメールを合わせて表示 実際にはうまくいかないことが多い
機能ゲノム学(第6回)
") バイオインフォマティクス次世代シーケンサー (NGS) 編 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 バイオインフォマティクス人材育成講座 スタンダードコース 2 自己紹介 1995 年 3 月 高知工業高等専門学校
バイオインフォマティクス次世代シーケンサー (NGS) 編 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 バイオインフォマティクス人材育成講座 スタンダードコース 2 自己紹介 1995 年 3 月 高知工業高等専門学校
※ ポイント ※
 4S-RO ロボティクス実験 参考資料 ファイル入出力 : ファイルの読み込み 1 周目に計測した生体情報データを読み込み プログラムにより信号処理を行うが その際にファイルの 入出力が必要となる 実験前半ですでに学習しているが必要に応じて本資料を参考にすること 以下のようにすると指定したファイルを読み込むことができる ( 詳細は後から記述 ) int i; double --------; char
4S-RO ロボティクス実験 参考資料 ファイル入出力 : ファイルの読み込み 1 周目に計測した生体情報データを読み込み プログラムにより信号処理を行うが その際にファイルの 入出力が必要となる 実験前半ですでに学習しているが必要に応じて本資料を参考にすること 以下のようにすると指定したファイルを読み込むことができる ( 詳細は後から記述 ) int i; double --------; char
KDDI ホスティングサービス G120 KDDI ホスティングサービス G200 WordPress インストールガイド ( ご参考資料 ) rev.1.2 KDDI 株式会社 1
 rev.1.2 KDDI 株式会社 1") KDDI ホスティングサービス G120 KDDI ホスティングサービス G200 WordPress インストールガイド ( ご参考資料 ) rev.1.2 KDDI 株式会社 1 ( 目次 ) 1. WordPress インストールガイド... 3 1-1 はじめに... 3 1-2 制限事項... 3 1-3 サイト初期設定... 4 2. WordPress のインストール ( コントロールパネル付属インストーラより
KDDI ホスティングサービス G120 KDDI ホスティングサービス G200 WordPress インストールガイド ( ご参考資料 ) rev.1.2 KDDI 株式会社 1 ( 目次 ) 1. WordPress インストールガイド... 3 1-1 はじめに... 3 1-2 制限事項... 3 1-3 サイト初期設定... 4 2. WordPress のインストール ( コントロールパネル付属インストーラより
ソフトウェア基礎 Ⅰ Report#2 提出日 : 2009 年 8 月 11 日 所属 : 工学部情報工学科 学籍番号 : K 氏名 : 當銘孔太
 ソフトウェア基礎 Ⅰ Report#2 提出日 : 2009 年 8 月 11 日 所属 : 工学部情報工学科 学籍番号 : 095739 K 氏名 : 當銘孔太 1. UNIX における正規表現とは何か, 使い方の例を挙げて説明しなさい. 1.1 正規表現とは? 正規表現 ( 正則表現ともいう ) とは ある規則に基づいて文字列 ( 記号列 ) の集合を表す方法の 1 つです ファイル名表示で使うワイルドカードも正規表現の兄弟みたいなもの
ソフトウェア基礎 Ⅰ Report#2 提出日 : 2009 年 8 月 11 日 所属 : 工学部情報工学科 学籍番号 : 095739 K 氏名 : 當銘孔太 1. UNIX における正規表現とは何か, 使い方の例を挙げて説明しなさい. 1.1 正規表現とは? 正規表現 ( 正則表現ともいう ) とは ある規則に基づいて文字列 ( 記号列 ) の集合を表す方法の 1 つです ファイル名表示で使うワイルドカードも正規表現の兄弟みたいなもの
GWB
 NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 参照ゲノム配列へのマッピング 再アライメント 変異検出 CLC Genomics Workbench または Biomedical Genomics Workbench
NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 参照ゲノム配列へのマッピング 再アライメント 変異検出 CLC Genomics Workbench または Biomedical Genomics Workbench
Microsoft Word - index.html
 R のインストールと超入門 R とは? R ダウンロード R のインストール R の基本操作 R 終了データの読み込みパッケージの操作 R とは? Rはデータ解析 マイニングを行うフリーソフトである Rはデータ解析の環境でもあり 言語でもある ニュージーランドのオークランド (Auckland) 大学の統計学科のRobert Gentlemanと Ross Ihakaにより開発がはじめられ 1997
R のインストールと超入門 R とは? R ダウンロード R のインストール R の基本操作 R 終了データの読み込みパッケージの操作 R とは? Rはデータ解析 マイニングを行うフリーソフトである Rはデータ解析の環境でもあり 言語でもある ニュージーランドのオークランド (Auckland) 大学の統計学科のRobert Gentlemanと Ross Ihakaにより開発がはじめられ 1997
Shareresearchオンラインマニュアル
 Chrome の初期設定 以下の手順で設定してください 1. ポップアップブロックの設定 2. 推奨する文字サイズの設定 3. 規定のブラウザに設定 4. ダウンロードファイルの保存先の設定 5.PDFレイアウトの印刷設定 6. ランキングやハイライトの印刷設定 7. 注意事項 なお 本マニュアルの内容は バージョン 61.0.3163.79 の Chrome を基に説明しています Chrome の設定手順や画面については
Chrome の初期設定 以下の手順で設定してください 1. ポップアップブロックの設定 2. 推奨する文字サイズの設定 3. 規定のブラウザに設定 4. ダウンロードファイルの保存先の設定 5.PDFレイアウトの印刷設定 6. ランキングやハイライトの印刷設定 7. 注意事項 なお 本マニュアルの内容は バージョン 61.0.3163.79 の Chrome を基に説明しています Chrome の設定手順や画面については
Maser - User Operation Manual
 Maser 3 Cell Innovation User Operation Manual 2013.4.1 1 目次 1. はじめに... 3 1.1. 推奨動作環境... 3 2. データの登録... 4 2.1. プロジェクトの作成... 4 2.2. Projectへのデータのアップロード... 8 2.2.1. HTTPSでのアップロード... 8 2.2.2. SFTPでのアップロード...
Maser 3 Cell Innovation User Operation Manual 2013.4.1 1 目次 1. はじめに... 3 1.1. 推奨動作環境... 3 2. データの登録... 4 2.1. プロジェクトの作成... 4 2.2. Projectへのデータのアップロード... 8 2.2.1. HTTPSでのアップロード... 8 2.2.2. SFTPでのアップロード...
日本乳酸菌学会誌の連載第11回ウェブ資料
 207.0.02 版 次世代シーケンサーデータの解析手法第 回統合データ解析環境 Galaxy ウェブ資料 大田達郎 * 寺田朋子 清水謙多郎 門田幸二 * 日本乳酸菌学会誌の連載第 回 W:Google Scholar Galaxy の 2005 年の論文の引用回数は 2,467 回 2 Giardine et al., Genome Res., 5: 45-455, 2005 日本乳酸菌学会誌の連載第
207.0.02 版 次世代シーケンサーデータの解析手法第 回統合データ解析環境 Galaxy ウェブ資料 大田達郎 * 寺田朋子 清水謙多郎 門田幸二 * 日本乳酸菌学会誌の連載第 回 W:Google Scholar Galaxy の 2005 年の論文の引用回数は 2,467 回 2 Giardine et al., Genome Res., 5: 45-455, 2005 日本乳酸菌学会誌の連載第
Microsoft Word - 1 color Normalization Document _Agilent version_ .doc
 color 実験の Normalization color 実験で得られた複数のアレイデータを相互比較するためには Normalization( 正規化 ) が必要です 2 つのサンプルを異なる色素でラベル化し 競合ハイブリダイゼーションさせる 2color 実験では 基本的に Dye Normalization( 色素補正 ) が適用されますが color 実験では データの特徴と実験の目的 (
color 実験の Normalization color 実験で得られた複数のアレイデータを相互比較するためには Normalization( 正規化 ) が必要です 2 つのサンプルを異なる色素でラベル化し 競合ハイブリダイゼーションさせる 2color 実験では 基本的に Dye Normalization( 色素補正 ) が適用されますが color 実験では データの特徴と実験の目的 (
1. 画面のリンクやボタン タブをクリックしても反応しない 現象 ログイン後 リンクやタブをクリックしても反応がない ( 画面が変わらない ) 原因 (1)Internet Explorerの互換表示設定が外れている (2) ツールバーの アドオン と呼ばれる追加機能によりブロックされている (1)
 原因 (1)Internet Explorerの互換表示設定が外れている (2) ツールバーの アドオン と呼ばれる追加機能によりブロックされている (1)") 最近よくあるお問い合わせ 本マニュアルでは 最近よくあるお問い合わせの解決手順をまとめました また 手順が難しいものについては 映像マニュアル をご用意しました 以下より 該当する現象を選択してください 2013.5.20 改定 ver. 目次 1. 画面のリンクやボタン タブをクリックしても反応しない 2 2. ライセンス認証に失敗 受講画面が準備完了のまま 受講画面が真っ白 4 3. Windows8
最近よくあるお問い合わせ 本マニュアルでは 最近よくあるお問い合わせの解決手順をまとめました また 手順が難しいものについては 映像マニュアル をご用意しました 以下より 該当する現象を選択してください 2013.5.20 改定 ver. 目次 1. 画面のリンクやボタン タブをクリックしても反応しない 2 2. ライセンス認証に失敗 受講画面が準備完了のまま 受講画面が真っ白 4 3. Windows8
Microsoft PowerPoint - prog03.ppt
 プログラミング言語 3 第 03 回 (2007 年 10 月 08 日 ) 1 今日の配布物 片面の用紙 1 枚 今日の課題が書かれています 本日の出欠を兼ねています 2/33 今日やること http://www.tnlab.ice.uec.ac.jp/~s-okubo/class/java06/ にアクセスすると 教材があります 2007 年 10 月 08 日分と書いてある部分が 本日の教材です
プログラミング言語 3 第 03 回 (2007 年 10 月 08 日 ) 1 今日の配布物 片面の用紙 1 枚 今日の課題が書かれています 本日の出欠を兼ねています 2/33 今日やること http://www.tnlab.ice.uec.ac.jp/~s-okubo/class/java06/ にアクセスすると 教材があります 2007 年 10 月 08 日分と書いてある部分が 本日の教材です
スライド 1
 適用マニュアル Hos-CanR 3.0 サービスパック適用マニュアル システム管理者用 SP1.4 バージョン 改訂日付 改訂内容 SP 1.4 2011/12/05 SP1.4リリースに伴う修正 SP 1.3 2011/11/01 リリースに伴う修正 SP 1.2 2010/12/10 SP1.2リリースに伴う修正 SP 1.1 2010/08/09 SP1.1リリースに伴う修正 SP 1.0
適用マニュアル Hos-CanR 3.0 サービスパック適用マニュアル システム管理者用 SP1.4 バージョン 改訂日付 改訂内容 SP 1.4 2011/12/05 SP1.4リリースに伴う修正 SP 1.3 2011/11/01 リリースに伴う修正 SP 1.2 2010/12/10 SP1.2リリースに伴う修正 SP 1.1 2010/08/09 SP1.1リリースに伴う修正 SP 1.0
nagasaki_GMT2015_key09
 Workflow Variant Calling 03 長崎は遺伝研 大量遺伝情報研究室の所属です 国立遺伝学研究所 生命情報研究センター 3F 2F 欧州EBIと米国NCBIと密接に協力しながら DDBJ/EMBL/GenBank国際塩基配列データ ベースを構築しています 私たちは 塩基配列登録を支援するシステムづくり 登録データを活用するシステムづくり 高速シーケンス配列の情報解析 を行なっています
Workflow Variant Calling 03 長崎は遺伝研 大量遺伝情報研究室の所属です 国立遺伝学研究所 生命情報研究センター 3F 2F 欧州EBIと米国NCBIと密接に協力しながら DDBJ/EMBL/GenBank国際塩基配列データ ベースを構築しています 私たちは 塩基配列登録を支援するシステムづくり 登録データを活用するシステムづくり 高速シーケンス配列の情報解析 を行なっています
PowerPoint Presentation
 エピジェノミクス解析編 2016/08/10 Filgen ChIP-seq (Transfactor & Histone), Bisulfite webex seminar 株式会社キアゲンアプライドアドバンストゲノミクス宮本真理, PhD 1 アジェンダ ChIP-seq 解析 Transcription Factor ChIP-seq Histone ChIP-seq Bisulfite-seq
エピジェノミクス解析編 2016/08/10 Filgen ChIP-seq (Transfactor & Histone), Bisulfite webex seminar 株式会社キアゲンアプライドアドバンストゲノミクス宮本真理, PhD 1 アジェンダ ChIP-seq 解析 Transcription Factor ChIP-seq Histone ChIP-seq Bisulfite-seq
1 開発ツールのインストール 最初に JDK をインストールし 次に IDE をインストールする という手順になります 1. JDK のインストール JDK のダウンロードとインストール JDK は次の URL でオラクル社のウェブページからダウンロードします
 1 開発ツールのインストール 最初に JDK をインストールし 次に IDE をインストールする という手順になります 1. JDK のインストール JDK のダウンロードとインストール JDK は次の URL でオラクル社のウェブページからダウンロードします http://www.oracle.com/technetwork/java/javase/downloads/index.html なお
1 開発ツールのインストール 最初に JDK をインストールし 次に IDE をインストールする という手順になります 1. JDK のインストール JDK のダウンロードとインストール JDK は次の URL でオラクル社のウェブページからダウンロードします http://www.oracle.com/technetwork/java/javase/downloads/index.html なお
本書について 本書では Winmostar を初めて使う人を対象に その導入手順と基本操作を紹介します 不明な点がある場合や本書の通りに動かない場合はまず 随時更新されている よくある質問 をご確認ください 2
 ビギナーズガイド 2018 年 10 月 5 日 株式会社クロスアビリティ 本書について 本書では Winmostar を初めて使う人を対象に その導入手順と基本操作を紹介します 不明な点がある場合や本書の通りに動かない場合はまず 随時更新されている よくある質問 https://winmostar.com/jp/qa_jp.php をご確認ください 2 Winmostar とは Winmostar
ビギナーズガイド 2018 年 10 月 5 日 株式会社クロスアビリティ 本書について 本書では Winmostar を初めて使う人を対象に その導入手順と基本操作を紹介します 不明な点がある場合や本書の通りに動かない場合はまず 随時更新されている よくある質問 https://winmostar.com/jp/qa_jp.php をご確認ください 2 Winmostar とは Winmostar
目次 専用アプリケーションをインストールする 1 アカウントを設定する 5 Windows クライアントから利用できる機能の紹介 7 1ファイル フォルダのアップロードとダウンロード 8 2ファイル更新履歴の管理 10 3 操作履歴の確認 12 4アクセスチケットの生成 ( フォルダ / ファイルの
 ServersMan@Disk Windows 版専用アプリケーション操作マニュアル 目次 専用アプリケーションをインストールする 1 アカウントを設定する 5 Windows クライアントから利用できる機能の紹介 7 1ファイル フォルダのアップロードとダウンロード 8 2ファイル更新履歴の管理 10 3 操作履歴の確認 12 4アクセスチケットの生成 ( フォルダ / ファイルの公開 ) 13
ServersMan@Disk Windows 版専用アプリケーション操作マニュアル 目次 専用アプリケーションをインストールする 1 アカウントを設定する 5 Windows クライアントから利用できる機能の紹介 7 1ファイル フォルダのアップロードとダウンロード 8 2ファイル更新履歴の管理 10 3 操作履歴の確認 12 4アクセスチケットの生成 ( フォルダ / ファイルの公開 ) 13
PowerPoint プレゼンテーション
 最近よくあるお問い合わせ 本マニュアルでは 最近よくあるお問い合わせの解決手順をまとめました 以下より 該当する現象を選択してください 2014.4.30 改定 ver. 目次 0. 必ずお読み下さい ユーザサポートツールが新しくなりました 2 1. 画面のリンクやボタン タブをクリックしても反応しない 3 2. ライセンス認証に失敗 受講画面が準備完了のまま 受講画面が真っ白 7 3. Windows8
最近よくあるお問い合わせ 本マニュアルでは 最近よくあるお問い合わせの解決手順をまとめました 以下より 該当する現象を選択してください 2014.4.30 改定 ver. 目次 0. 必ずお読み下さい ユーザサポートツールが新しくなりました 2 1. 画面のリンクやボタン タブをクリックしても反応しない 3 2. ライセンス認証に失敗 受講画面が準備完了のまま 受講画面が真っ白 7 3. Windows8
( 目次 ) 1. XOOPSインストールガイド はじめに 制限事項 サイト初期設定 XOOPSのインストール はじめに データベースの作成 XOOPSのインストール
 1. XOOPSインストールガイド はじめに 制限事項 サイト初期設定 XOOPSのインストール はじめに データベースの作成 XOOPSのインストール") KDDI ホスティングサービス (G120, G200) XOOPS インストールガイド ( ご参考資料 ) rev1.0 KDDI 株式会社 1 ( 目次 ) 1. XOOPSインストールガイド...3 1-1 はじめに...3 1-2 制限事項...3 1-3 サイト初期設定...4 2. XOOPSのインストール...9 3-1 はじめに...9 3-2 データベースの作成...9 3-3 XOOPSのインストール...10
KDDI ホスティングサービス (G120, G200) XOOPS インストールガイド ( ご参考資料 ) rev1.0 KDDI 株式会社 1 ( 目次 ) 1. XOOPSインストールガイド...3 1-1 はじめに...3 1-2 制限事項...3 1-3 サイト初期設定...4 2. XOOPSのインストール...9 3-1 はじめに...9 3-2 データベースの作成...9 3-3 XOOPSのインストール...10
CLC Genomics Workbench ウェブトレーニングセミナー: 変異解析編
 CLC Genomics Workbench ウェブトレーニングセミナー : 遺伝子発現解析編 12 th Feb., 2016 フィルジェン株式会社バイオサイエンス部 biosupport@filgen.jp Feb., 2016_V2 1 遺伝子発現解析概要 本日のセミナーにおける解析の流れ及び使用するツール名 ( 図中赤枠部分 ) Case Control インポート インポート インポート
CLC Genomics Workbench ウェブトレーニングセミナー : 遺伝子発現解析編 12 th Feb., 2016 フィルジェン株式会社バイオサイエンス部 biosupport@filgen.jp Feb., 2016_V2 1 遺伝子発現解析概要 本日のセミナーにおける解析の流れ及び使用するツール名 ( 図中赤枠部分 ) Case Control インポート インポート インポート
厚生労働省版ストレスチェック実施プログラム 設置 設定マニュアル Ver.3.0 目次 1. プログラム概要 設置手順 注意事項 動作環境 初期設定 ( 環境設定 ) 初期設定 ( パスワード変更 ) 初
 初期設定 ( パスワード変更 ) 初") 厚生労働省版ストレスチェック実施プログラム 設置 設定マニュアル Ver.3.0 目次 1. プログラム概要... 2 2. 設置手順... 3 3. 注意事項... 5 4. 動作環境... 6 5. 初期設定 ( 環境設定 )... 7 6. 初期設定 ( パスワード変更 )... 9 7. 初期設定 ( 面接指導医登録 )... 11 8. 初期設定 ( 実施設定 )... 12 9. 初期設定
厚生労働省版ストレスチェック実施プログラム 設置 設定マニュアル Ver.3.0 目次 1. プログラム概要... 2 2. 設置手順... 3 3. 注意事項... 5 4. 動作環境... 6 5. 初期設定 ( 環境設定 )... 7 6. 初期設定 ( パスワード変更 )... 9 7. 初期設定 ( 面接指導医登録 )... 11 8. 初期設定 ( 実施設定 )... 12 9. 初期設定
I
 別紙 LG-WAN 決算統計データの操作手順 LG-WANに掲載されている決算統計データをワークシート H24(2012)~S44(1969) の所定の場所にデータを貼り付けることにより 貸借対照表の 有形固定資産 に数値が反映されます LG-WAN 決算統計データは 調査データ閲覧 ダウンロード のページに掲載されています URL: http://llb.k3tokei.asp.lgwan.jp/soumu-app/contents/index.html
別紙 LG-WAN 決算統計データの操作手順 LG-WANに掲載されている決算統計データをワークシート H24(2012)~S44(1969) の所定の場所にデータを貼り付けることにより 貸借対照表の 有形固定資産 に数値が反映されます LG-WAN 決算統計データは 調査データ閲覧 ダウンロード のページに掲載されています URL: http://llb.k3tokei.asp.lgwan.jp/soumu-app/contents/index.html
厚生労働省版ストレスチェック実施プログラムバージョンアップマニュアル (Ver2.2 から Ver.3.2) 目次 1. プログラム概要 バージョンアップ実施手順 要注意 zip ファイル解凍の準備 Windows によって PC が保護されました と
 目次 1. プログラム概要 バージョンアップ実施手順 要注意 zip ファイル解凍の準備 Windows によって PC が保護されました と") 厚生労働省版ストレスチェック実施プログラムバージョンアップマニュアル (Ver2.2 から Ver.3.2) 目次 1. プログラム概要... 2 2. バージョンアップ実施手順... 3 3. 要注意 zip ファイル解凍の準備... 9 4. Windows によって PC が保護されました というダイアログが表示される場合.. 10 5. 初回実行時にインストーラが実行される場合... 11
厚生労働省版ストレスチェック実施プログラムバージョンアップマニュアル (Ver2.2 から Ver.3.2) 目次 1. プログラム概要... 2 2. バージョンアップ実施手順... 3 3. 要注意 zip ファイル解凍の準備... 9 4. Windows によって PC が保護されました というダイアログが表示される場合.. 10 5. 初回実行時にインストーラが実行される場合... 11
 GenBank クイックスタート GenBank は NLM/NCBI にて維持管理されている核酸配列データベースです また GenBank は EMBL, DDBJ と三極間で連携しながら国際核酸配列データベースを共同で構築しています これら三機関はデータを日々交換し続けており その規模は 160000 種にも及ぶ生物種の塩基配列から成り立つまでになっています この GenBank クイックスタートでは
GenBank クイックスタート GenBank は NLM/NCBI にて維持管理されている核酸配列データベースです また GenBank は EMBL, DDBJ と三極間で連携しながら国際核酸配列データベースを共同で構築しています これら三機関はデータを日々交換し続けており その規模は 160000 種にも及ぶ生物種の塩基配列から成り立つまでになっています この GenBank クイックスタートでは
クライアント証明書インストールマニュアル
 事前設定付クライアント証明書インストールマニュアル このマニュアルは クライアント証明書インストールマニュアル の手順で証明書がインストールできなかった方のための インストールマニュアルです エクストラネットは Internet Explorer をご利用ください Microsoft Edge 他 Internet Explorer 以外のブラウザではご利用になれません 当マニュアル利用にあたっては
事前設定付クライアント証明書インストールマニュアル このマニュアルは クライアント証明書インストールマニュアル の手順で証明書がインストールできなかった方のための インストールマニュアルです エクストラネットは Internet Explorer をご利用ください Microsoft Edge 他 Internet Explorer 以外のブラウザではご利用になれません 当マニュアル利用にあたっては
誓約書の同意 4 初回のみ 下記画面が表示されるので内容を確認後 同意する ボタンをクリック 同意していただけない場合はネット調達システムを使うことができません 参照条件設定 5 案件の絞り込み画面が表示されます 5-1 施工地域を選択して 施工地域選択完了 ボタンをクリック - 2 -
 ネット調達システム簡易マニュアル 協力会社編 システムの起動 ~ 案件参照 ~ 見積提出 ログイン 1OC-COMET にログインします 2 左側のメニューより 関連システム連携 ( 見積回答 S 他 ) をクリック 3 ネット調達システム をクリック - 1 - 誓約書の同意 4 初回のみ 下記画面が表示されるので内容を確認後 同意する ボタンをクリック 同意していただけない場合はネット調達システムを使うことができません
ネット調達システム簡易マニュアル 協力会社編 システムの起動 ~ 案件参照 ~ 見積提出 ログイン 1OC-COMET にログインします 2 左側のメニューより 関連システム連携 ( 見積回答 S 他 ) をクリック 3 ネット調達システム をクリック - 1 - 誓約書の同意 4 初回のみ 下記画面が表示されるので内容を確認後 同意する ボタンをクリック 同意していただけない場合はネット調達システムを使うことができません
JavaScriptで プログラミング
 JavaScript でプログラミング JavaScript とは プログラミング言語の 1 つ Web ページ上でプログラムを動かすことが主目的 Web ブラウザで動かすことができる 動作部分の書き方が C や Java などに似ている 2 JavaScript プログラムを動かすには の範囲を 1. テキストエディタで入力 2..html というファイル名で保存
JavaScript でプログラミング JavaScript とは プログラミング言語の 1 つ Web ページ上でプログラムを動かすことが主目的 Web ブラウザで動かすことができる 動作部分の書き方が C や Java などに似ている 2 JavaScript プログラムを動かすには の範囲を 1. テキストエディタで入力 2..html というファイル名で保存
Microsoft Word A02
 1 / 10 ページ キャリアアップコンピューティング 第 2 講 [ 全 15 講 ] 2018 年度 2 / 10 ページ 第 2 講ビジネスドキュメントの基本 2-1 Word の起動 画面構成 Word を起動し 各部の名称と機能を確認してみましょう 2 1 3 6 4 5 名称 機能 1 タイトルバー アプリケーション名とファイル名が表示されます 2 クイックアクセスツールバー よく使うコマンドを登録できます
1 / 10 ページ キャリアアップコンピューティング 第 2 講 [ 全 15 講 ] 2018 年度 2 / 10 ページ 第 2 講ビジネスドキュメントの基本 2-1 Word の起動 画面構成 Word を起動し 各部の名称と機能を確認してみましょう 2 1 3 6 4 5 名称 機能 1 タイトルバー アプリケーション名とファイル名が表示されます 2 クイックアクセスツールバー よく使うコマンドを登録できます
Microsoft Word - _ ‘C’³_V1.6InstManual.doc
 厚生労働省版医薬品等電子申請ソフト ダウンロードマニュアル 平成 23 年 6 月 17 日版 厚生労働省医薬食品局 はじめに 本マニュアルの利用方法 本マニュアルは 医薬品等電子申請ソフト ( 以下 申請ソフト と呼びます ) のダウンロード方法について説明したものです 申請ソフトのダウンロードは ウェブブラウザを用いたインターネットからの一般のドキュメントやプログラムのダウンロードと大きく異なるところはありません
厚生労働省版医薬品等電子申請ソフト ダウンロードマニュアル 平成 23 年 6 月 17 日版 厚生労働省医薬食品局 はじめに 本マニュアルの利用方法 本マニュアルは 医薬品等電子申請ソフト ( 以下 申請ソフト と呼びます ) のダウンロード方法について説明したものです 申請ソフトのダウンロードは ウェブブラウザを用いたインターネットからの一般のドキュメントやプログラムのダウンロードと大きく異なるところはありません
鳥取県物品電子入札システムセキュリティ ポリシー設定マニュアル IC カードを利用しない応札者向け 第 1.7 版 平成 31 年 2 月鳥取県物品契約課 鳥取県物品電子入札システムセキュリティ ポリシー設定マニュアル Ver.01-07
 鳥取県物品電子入札システムセキュリティ ポリシー設定マニュアル IC カードを利用しない応札者向け 第 1.7 版 平成 31 年 2 月鳥取県物品契約課 目次 第 1 章はじめに 1 1.1 セキュリティ ポリシーを設定するまでの流れ 1 第 2 章セキュリティ ポリシーを設定する前に 2 2.1 前提条件確認 2 2.2 Java ランタイム (JRE) の確認方法 3 第 3 章 Java 実行環境の設定
鳥取県物品電子入札システムセキュリティ ポリシー設定マニュアル IC カードを利用しない応札者向け 第 1.7 版 平成 31 年 2 月鳥取県物品契約課 目次 第 1 章はじめに 1 1.1 セキュリティ ポリシーを設定するまでの流れ 1 第 2 章セキュリティ ポリシーを設定する前に 2 2.1 前提条件確認 2 2.2 Java ランタイム (JRE) の確認方法 3 第 3 章 Java 実行環境の設定
Rでゲノム・トランスクリプトーム解析
 06.08. 版 スライド 8 までは自習 当日はスライド 9 から始める予定 スライド 3-86 は当日省略予定 講習会後に各自で復習してください 第 3 部 :NGS 解析 ( 中 ~ 上級 ) ~ トランスクリプトームアセンブリ 発現量推定 ~ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
06.08. 版 スライド 8 までは自習 当日はスライド 9 から始める予定 スライド 3-86 は当日省略予定 講習会後に各自で復習してください 第 3 部 :NGS 解析 ( 中 ~ 上級 ) ~ トランスクリプトームアセンブリ 発現量推定 ~ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
PowerPoint プレゼンテーション
 最近よくあるお問い合わせ 本マニュアルでは 最近よくあるお問い合わせの解決手順をまとめました 以下より 該当する現象を選択してください 2014.6.22 改定 ver. 目次 1. トップページの各メニューをクリックしても反応が無い 3 2. 動画再生画面が真っ白になる 7 3. 準備完了 と表示されたまま動画再生が始まらない 13 4. このファイルを再生する権限はありません のメッセージが表示され動画再生が始まらない
最近よくあるお問い合わせ 本マニュアルでは 最近よくあるお問い合わせの解決手順をまとめました 以下より 該当する現象を選択してください 2014.6.22 改定 ver. 目次 1. トップページの各メニューをクリックしても反応が無い 3 2. 動画再生画面が真っ白になる 7 3. 準備完了 と表示されたまま動画再生が始まらない 13 4. このファイルを再生する権限はありません のメッセージが表示され動画再生が始まらない
目次 第 1 章はじめに 電子入札システムを使用するまでの流れ 1 第 2 章 Java ポリシーを設定する前に 前提条件の確認 2 第 3 章 Java のバージョンについて Java バージョン確認方法 Java のアンインストール ( ケース2の
 電子入札サービス IC カードを利用しない事業者向け Java ポリシー設定マニュアル (Windows10 用 ) 平成 28 年 6 月 目次 第 1 章はじめに 1 1.1 電子入札システムを使用するまでの流れ 1 第 2 章 Java ポリシーを設定する前に 2 2.1 前提条件の確認 2 第 3 章 Java のバージョンについて 4 3.1 Java バージョン確認方法 4 3.2 Java
電子入札サービス IC カードを利用しない事業者向け Java ポリシー設定マニュアル (Windows10 用 ) 平成 28 年 6 月 目次 第 1 章はじめに 1 1.1 電子入札システムを使用するまでの流れ 1 第 2 章 Java ポリシーを設定する前に 2 2.1 前提条件の確認 2 第 3 章 Java のバージョンについて 4 3.1 Java バージョン確認方法 4 3.2 Java
スライド 1
 Hos-CanR 2.5 3.0 クライアント サーバー (CS) 版データ移行マニュアル Hos-CanR クライアント サーバー (CS) 版 Ver. 2.5 Ver. 3.0 データ移行マニュアル システム管理者用 Ver. 2 バージョン改訂日付改訂内容 Ver. 1 2010/3/15 初版 Ver. 2 2010/12/10 作業対象コンピュータのアイコン追加 Hos-CanR 2.5
Hos-CanR 2.5 3.0 クライアント サーバー (CS) 版データ移行マニュアル Hos-CanR クライアント サーバー (CS) 版 Ver. 2.5 Ver. 3.0 データ移行マニュアル システム管理者用 Ver. 2 バージョン改訂日付改訂内容 Ver. 1 2010/3/15 初版 Ver. 2 2010/12/10 作業対象コンピュータのアイコン追加 Hos-CanR 2.5
分析のステップ Step 1: Y( 目的変数 ) に対する値の順序を確認 Step 2: モデルのあてはめ を実行 適切なモデルの指定 Step 3: オプションを指定し オッズ比とその信頼区間を表示 以下 このステップに沿って JMP の操作をご説明します Step 1: Y( 目的変数 ) の
 に対する値の順序を確認 Step 2: モデルのあてはめ を実行 適切なモデルの指定 Step 3: オプションを指定し オッズ比とその信頼区間を表示 以下 このステップに沿って JMP の操作をご説明します Step 1: Y( 目的変数 ) の") JMP によるオッズ比 リスク比 ( ハザード比 ) の算出と注意点 SAS Institute Japan 株式会社 JMP ジャパン事業部 2011 年 10 月改定 1. はじめに 本文書は JMP でロジスティック回帰モデルによるオッズ比 比例ハザードモデルによるリスク比 それぞれに対する信頼区間を求める操作方法と注意点を述べたものです 本文書は JMP 7 以降のバージョンに対応しております
JMP によるオッズ比 リスク比 ( ハザード比 ) の算出と注意点 SAS Institute Japan 株式会社 JMP ジャパン事業部 2011 年 10 月改定 1. はじめに 本文書は JMP でロジスティック回帰モデルによるオッズ比 比例ハザードモデルによるリスク比 それぞれに対する信頼区間を求める操作方法と注意点を述べたものです 本文書は JMP 7 以降のバージョンに対応しております
( 目次 ) 1. Joomla! インストールガイド はじめに 制限事項 サイト初期設定 Joomla! のインストール はじめに データベースの作成 Joomla! のインストール...
 1. Joomla! インストールガイド はじめに 制限事項 サイト初期設定 Joomla! のインストール はじめに データベースの作成 Joomla! のインストール...") KDDI ホスティングサービス (G120, G200) Joomla! インストールガイド ( ご参考資料 ) rev.1.0 KDDI 株式会社 1 ( 目次 ) 1. Joomla! インストールガイド...3 1-1 はじめに...3 1-2 制限事項...3 1-3 サイト初期設定...4 2. Joomla! のインストール...9 2-1 はじめに...9 2-2 データベースの作成...9
KDDI ホスティングサービス (G120, G200) Joomla! インストールガイド ( ご参考資料 ) rev.1.0 KDDI 株式会社 1 ( 目次 ) 1. Joomla! インストールガイド...3 1-1 はじめに...3 1-2 制限事項...3 1-3 サイト初期設定...4 2. Joomla! のインストール...9 2-1 はじめに...9 2-2 データベースの作成...9
もくじ 1. 全国統一小学生テストのシステムをご利用になる前に PC の初期設定について P3 2. よくあるご質問 (1) 生徒の申し込みについて (2) 実施要項について (3) 受験票 QRコードの印刷について (4) スキャン送信について (5) 実施後報告について P4 P5 P6 P7
 生徒の申し込みについて (2) 実施要項について (3) 受験票 QRコードの印刷について (4) スキャン送信について (5) 実施後報告について P4 P5 P6 P7") Q&A 集 全国統一小学生テスト実行委員会 もくじ 1. 全国統一小学生テストのシステムをご利用になる前に PC の初期設定について P3 2. よくあるご質問 (1) 生徒の申し込みについて (2) 実施要項について (3) 受験票 QRコードの印刷について (4) スキャン送信について (5) 実施後報告について P4 P5 P6 P7 P8 3. お問い合わせ先 P10 2 1. 全国統一小学生テストのシステムをご利用になる前に
Q&A 集 全国統一小学生テスト実行委員会 もくじ 1. 全国統一小学生テストのシステムをご利用になる前に PC の初期設定について P3 2. よくあるご質問 (1) 生徒の申し込みについて (2) 実施要項について (3) 受験票 QRコードの印刷について (4) スキャン送信について (5) 実施後報告について P4 P5 P6 P7 P8 3. お問い合わせ先 P10 2 1. 全国統一小学生テストのシステムをご利用になる前に
シヤチハタ デジタルネーム 操作マニュアル
 操作マニュアル 目次 1 はじめに... 2 2 動作環境... 2 3 インストール... 3 4 印鑑を登録する... 6 5 登録した印鑑を削除する... 9 6 印鑑を捺印する... 10 6.1 Word 文書へ捺印する... 10 6.2 Excel 文書へ捺印する... 12 7 コピー & ペーストで捺印する... 13 8 印鑑の色を変更する... 15 9 印鑑の順番を入れ替える...
操作マニュアル 目次 1 はじめに... 2 2 動作環境... 2 3 インストール... 3 4 印鑑を登録する... 6 5 登録した印鑑を削除する... 9 6 印鑑を捺印する... 10 6.1 Word 文書へ捺印する... 10 6.2 Excel 文書へ捺印する... 12 7 コピー & ペーストで捺印する... 13 8 印鑑の色を変更する... 15 9 印鑑の順番を入れ替える...
農業・農村基盤図の大字小字コードXML作成 説明書
 農業 農村基盤図の大字小字コード XML 作成説明書 2007/06/06 有限会社ジオ コーチ システムズ http://www.geocoach.co.jp/ info@geocoach.co.jp 農業 農村基盤図の大字小字コード XML 作成 プログラムについての説明書です バージョン ビルド 1.01 2007/06/06 農業 農村基盤図の大字小字コード XML 作成 は 市区町村 大字
農業 農村基盤図の大字小字コード XML 作成説明書 2007/06/06 有限会社ジオ コーチ システムズ http://www.geocoach.co.jp/ info@geocoach.co.jp 農業 農村基盤図の大字小字コード XML 作成 プログラムについての説明書です バージョン ビルド 1.01 2007/06/06 農業 農村基盤図の大字小字コード XML 作成 は 市区町村 大字
C プログラミング演習 1( 再 ) 2 講義では C プログラミングの基本を学び 演習では やや実践的なプログラミングを通して学ぶ
 2 講義では C プログラミングの基本を学び 演習では やや実践的なプログラミングを通して学ぶ") C プログラミング演習 1( 再 ) 2 講義では C プログラミングの基本を学び 演習では やや実践的なプログラミングを通して学ぶ 今回のプログラミングの課題 次のステップによって 徐々に難易度の高いプログラムを作成する ( 参照用の番号は よくわかる C 言語 のページ番号 ) 1. キーボード入力された整数 10 個の中から最大のものを答える 2. 整数を要素とする配列 (p.57-59) に初期値を与えておき
C プログラミング演習 1( 再 ) 2 講義では C プログラミングの基本を学び 演習では やや実践的なプログラミングを通して学ぶ 今回のプログラミングの課題 次のステップによって 徐々に難易度の高いプログラムを作成する ( 参照用の番号は よくわかる C 言語 のページ番号 ) 1. キーボード入力された整数 10 個の中から最大のものを答える 2. 整数を要素とする配列 (p.57-59) に初期値を与えておき
PowerPoint プレゼンテーション
 バイオインフォマティクス 講習会 V 事前準備 が完了されている方は コンテナの起動 ファイルのコピー (Windows) まで 進めておいてください メニュー 1. 環境構築の確認 2. 基本的なLinuxコマンド 3. ツールのインストール 4. NGSデータの基礎知識と前処理 5. トランスクリプトのアッセンブル 6. RNA-seqのリファレンスcDNAマッピングとFPKM 算出 7. RNA-seqのリファレンスゲノムマッピングとFPKM
バイオインフォマティクス 講習会 V 事前準備 が完了されている方は コンテナの起動 ファイルのコピー (Windows) まで 進めておいてください メニュー 1. 環境構築の確認 2. 基本的なLinuxコマンド 3. ツールのインストール 4. NGSデータの基礎知識と前処理 5. トランスクリプトのアッセンブル 6. RNA-seqのリファレンスcDNAマッピングとFPKM 算出 7. RNA-seqのリファレンスゲノムマッピングとFPKM
独立行政法人産業技術総合研究所 PMID-Extractor ユーザ利用マニュアル バイオメディシナル情報研究センター 2009/03/09 第 1.0 版
 独立行政法人産業技術総合研究所 PMID-Extractor ユーザ利用マニュアル バイオメディシナル情報研究センター 2009/03/09 第 1.0 版 目次 1. はじめに... 3 2. インストール方法... 4 3. プログラムの実行... 5 4. プログラムの終了... 5 5. 操作方法... 6 6. 画面の説明... 8 付録 A:Java のインストール方法について... 11
独立行政法人産業技術総合研究所 PMID-Extractor ユーザ利用マニュアル バイオメディシナル情報研究センター 2009/03/09 第 1.0 版 目次 1. はじめに... 3 2. インストール方法... 4 3. プログラムの実行... 5 4. プログラムの終了... 5 5. 操作方法... 6 6. 画面の説明... 8 付録 A:Java のインストール方法について... 11
ゲームプログラミング講習 第0章 導入
 ゲームプログラミング講習 第 0 章 導入 ゲーム制作に必要なものをインストールします ゲームプログラミング講習第 0 章導入 1 ゲーム制作に必要なもの Microsoft Visual Studio DXライブラリ プロジェクトテンプレート C 言語の知識 ゲームプログラミング講習第 0 章導入 2 Microsoft Visual Studio とは C 言語でプログラミングして Windows
ゲームプログラミング講習 第 0 章 導入 ゲーム制作に必要なものをインストールします ゲームプログラミング講習第 0 章導入 1 ゲーム制作に必要なもの Microsoft Visual Studio DXライブラリ プロジェクトテンプレート C 言語の知識 ゲームプログラミング講習第 0 章導入 2 Microsoft Visual Studio とは C 言語でプログラミングして Windows
リンクされたイメージを表示できません ファイルが移動または削除されたか 名前が変更された可能性があります リンクに正しいファイル名と場所が指定されていることを確認してください ここでは昨年までにいただいたご質問で多かったものについて その回答をまとめてあります 各種調査書様式の操作 Q12 調査書様
 Q1 パソコン基本設定 Windows Vista 又は Windows 7 は利用できますかご利用できます ただし Windows 8 については動作の確認がされていないため ご利用になれません また 2014 年 4 月をもって Windows XP のサポート期間が終了しております なるべくご利用はお控えください 編 Q2 Q3 Excel 2007 2010 2013 2016 は利用できますかご利用できます
Q1 パソコン基本設定 Windows Vista 又は Windows 7 は利用できますかご利用できます ただし Windows 8 については動作の確認がされていないため ご利用になれません また 2014 年 4 月をもって Windows XP のサポート期間が終了しております なるべくご利用はお控えください 編 Q2 Q3 Excel 2007 2010 2013 2016 は利用できますかご利用できます
ゲノム情報解析基礎 ~ Rで塩基配列解析 ~
 トランスクリプトーム解析の現況 ~ マイクロアレイ vs. RNA-seq~ 東京大学 大学院農学生命科学研究科 アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 スライド PDF はウェブから取得可能です 2 ステレオタイプなイメージ
トランスクリプトーム解析の現況 ~ マイクロアレイ vs. RNA-seq~ 東京大学 大学院農学生命科学研究科 アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 スライド PDF はウェブから取得可能です 2 ステレオタイプなイメージ
c3_op-manual
 研究者学術情報データベース操作説明 -- - 研究者 DB のログイン画面へのアクセス手順 () 立命館大学 トップページ http://www.ritsumei.ac.jp/ ドロップダウンメニューがスライドしてくるので 学内研究支援情報 ( 研究部 ) をクリック グローバルリンクの 研究 産学官連携 の文字にマウスポインタを合わせる クリック 次ページへ続く -- - 研究者 DB のログイン画面へのアクセス手順
研究者学術情報データベース操作説明 -- - 研究者 DB のログイン画面へのアクセス手順 () 立命館大学 トップページ http://www.ritsumei.ac.jp/ ドロップダウンメニューがスライドしてくるので 学内研究支援情報 ( 研究部 ) をクリック グローバルリンクの 研究 産学官連携 の文字にマウスポインタを合わせる クリック 次ページへ続く -- - 研究者 DB のログイン画面へのアクセス手順
Microsoft Word - 操作マニュアル-Excel-2.doc
 Excel プログラム開発の練習マニュアルー 1 ( 関数の学習 ) 作成 2015.01.31 修正 2015.02.04 本マニュアルでは Excel のプログラム開発を行なうに当たって まずは Excel の関数に関する学習 について記述する Ⅰ.Excel の関数に関する学習 1. 初めに Excel は単なる表計算のソフトと思っている方も多いと思います しかし Excel には 一般的に使用する
Excel プログラム開発の練習マニュアルー 1 ( 関数の学習 ) 作成 2015.01.31 修正 2015.02.04 本マニュアルでは Excel のプログラム開発を行なうに当たって まずは Excel の関数に関する学習 について記述する Ⅰ.Excel の関数に関する学習 1. 初めに Excel は単なる表計算のソフトと思っている方も多いと思います しかし Excel には 一般的に使用する
ご存知ですか? データ転送
 ご存知ですか? データ転送 System i のデータベースを PC にダウンロード System i 上のデータベースからデータを PC にダウンロードできます テキスト形式や CSV Excel(BIFF) 形式などに変換可能 System i データベースへのアップロードも可能 必要なライセンスプログラムは iseries Access for Windows(5722-XE1) または PCOMM
ご存知ですか? データ転送 System i のデータベースを PC にダウンロード System i 上のデータベースからデータを PC にダウンロードできます テキスト形式や CSV Excel(BIFF) 形式などに変換可能 System i データベースへのアップロードも可能 必要なライセンスプログラムは iseries Access for Windows(5722-XE1) または PCOMM
スライド 1
 ラベル屋さん HOME かんたんマニュアル リンクコース 目次 STEP 1-2 : ( 基礎編 ) 用紙の選択と文字の入力 STEP 3 : ( 基礎編 ) リンクの設定 STEP 4 : ( 基礎編 ) リンクデータの入力と印刷 STEP 5 : ( 応用編 ) リンクデータの入力 1 STEP 6 : ( 応用編 ) リンクデータの入力 2 STEP 7-8 : ( 応用編 ) リンク機能で使ったデータをコピーしたい場合
ラベル屋さん HOME かんたんマニュアル リンクコース 目次 STEP 1-2 : ( 基礎編 ) 用紙の選択と文字の入力 STEP 3 : ( 基礎編 ) リンクの設定 STEP 4 : ( 基礎編 ) リンクデータの入力と印刷 STEP 5 : ( 応用編 ) リンクデータの入力 1 STEP 6 : ( 応用編 ) リンクデータの入力 2 STEP 7-8 : ( 応用編 ) リンク機能で使ったデータをコピーしたい場合
<8B9E93738CF092CA904D94CC814090BF8B818F B D836A B B B816A2E786C73>
 京都交通信販 請求書 Web サービス操作マニュアル 第 9 版 (2011 年 2 月 1 日改訂 ) 京都交通信販株式会社 http://www.kyokoshin.co.jp TEL075-314-6251 FX075-314-6255 目次 STEP 1 >> ログイン画面 請求書 Web サービスログイン画面を確認します P.1 STEP 2 >> ログイン 請求書 Web サービスにログインします
京都交通信販 請求書 Web サービス操作マニュアル 第 9 版 (2011 年 2 月 1 日改訂 ) 京都交通信販株式会社 http://www.kyokoshin.co.jp TEL075-314-6251 FX075-314-6255 目次 STEP 1 >> ログイン画面 請求書 Web サービスログイン画面を確認します P.1 STEP 2 >> ログイン 請求書 Web サービスにログインします
ガイダンス
 情報科学 B 第 2 回変数 1 今日やること Java プログラムの書き方 変数とは何か? 2 Java プログラムの書き方 3 作業手順 Java 言語を用いてソースコードを記述する (Cpad エディタを使用 ) コンパイル (Cpad エディタを使用 ) 実行 (Cpad エディタを使用 ) エラーが出たらどうしたらよいか??? 4 書き方 これから作成する Hello.java 命令文 メソッドブロック
情報科学 B 第 2 回変数 1 今日やること Java プログラムの書き方 変数とは何か? 2 Java プログラムの書き方 3 作業手順 Java 言語を用いてソースコードを記述する (Cpad エディタを使用 ) コンパイル (Cpad エディタを使用 ) 実行 (Cpad エディタを使用 ) エラーが出たらどうしたらよいか??? 4 書き方 これから作成する Hello.java 命令文 メソッドブロック