Rでゲノム・トランスクリプトーム解析
|
|
|
- そうすけ こびき
- 4 years ago
- Views:
Transcription
1 R でゲノム トランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp 1
2 自己紹介 1995 年 3 月 高知工業高等専門学校 工業化学科卒業 1997 年 3 月 東京農工大学 工学部 物質生物工学科卒業 1999 年 3 月 東京農工大学 大学院工学研究科 物質生物工学専攻修士課程修了 2002 年 3 月 東京大学 大学院農学生命科学研究科 応用生命工学専攻博士課程修了 学位論文 : cdna マイクロアレイを用いた遺伝子発現解析手法の開発 ( 指導教官 : 清水謙多郎教授 ) 2002/4/1~ 産総研 生命情報科学研究センター (CBRC) 産総研特別研究員 2003/11/1~ 放医研 先端遺伝子発現研究センター研究員 2005/2/16~ 東京大学 大学院農学生命科学研究科特任助手 2

3 参考 URL 自前 PC でやる場合はここを参考にして必要なパッケージを予めインストールしておかねばなりません ( 数時間程度かかります ) 3
4 Contents(R で...) ゲノム解析 アノテーションファイルを読み込んで目的のキーワードを含む行のみ抽出 multi-fasta ファイルを自在に解析 配列長分布 GC 含量 フィルタリング 部分配列の切り出しなど 連続塩基の出現頻度 (CpG) 解析 ゲノム配列取得など トランスクリプトーム解析 研究目的別留意点 : サンプル内とサンプル間の違い マッピング カウント情報取得 データを眺める : クラスタリングや M-A plot 理想的な実験デザイン なぜ x 倍発現変動という議論がだめなんですか? モデルとか分布って 自分の解析結果にどういう影響を与えているの? 多重比較問題 :FDR って何? 4



5 アノテーションファイル?! アノテーショ 遺伝子ごとに どの染色体上に存在するのかやどんな Gene Ontology ID が割り当てられているのかなどの情報を含むファイル 5
6 アノテーションファイルからの情報抽出 核 (nucleus) に存在する遺伝子のみからなるリストを得たいときにも R が利用可能 6
7 アノテーションファイルからの情報抽出 入力 : アノテーションファイル (annotation.txt) 出力 :hoge1.txt 入力 : リストファイル (genelist1.txt) 目的 : アノテーションファイル (annotation.txt) 中の第 1 列目に対して リストファイル (genelist1.txt) 中の文字列と一致する行を抜き出して hoge1.txt というファイル名で出力したい 7
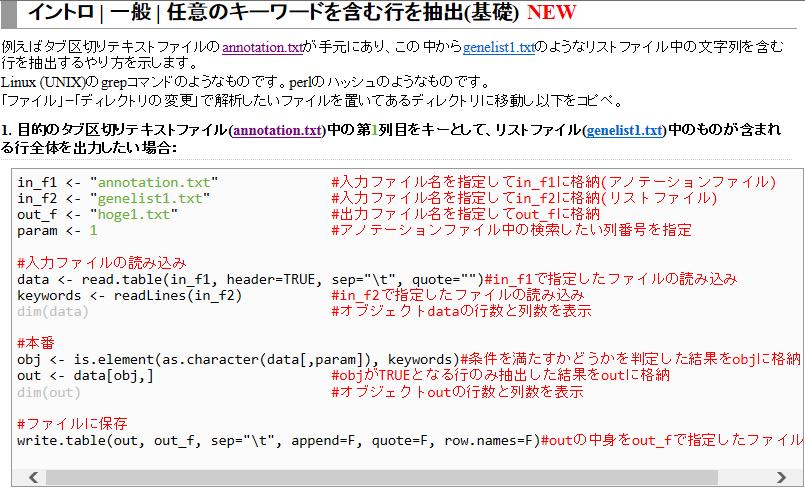
8 8

9 入力 1: annotation.txt 入力 2: genelist1.txt 出力 : hoge1.txt デスクトップ上に hoge という名前のフォルダがあり フォルダ中に annotation.txt と genelist1.txt が存在するという前提です メモ帳で開くと改行コードが崩れている場合は ワードパッドなどで開くとよい 9

10 R の起動 デスクトップにある hoge フォルダ中のファイルを解析 10



11 作業ディレクトリの変更
12 getwd() と打ち込んで確認 12


13 基本はコピペ 2013 年 7 月以降のリニューアルで コードのコピーがやりずらくなっています CTRL と ALT キーを押しながらコードの枠内で左クリックすると 全選択できます 1 一連のコマンド群をコピーして 2R Console 画面上でペースト 13


14 実行結果 実行前の hoge フォルダ 実行後の hoge フォルダ 14
15 色についての説明 15



16 色についての説明 上記は 1 列目でキーワード検索する場合 4 列目でキーワード検索したいときは? 16



17 解答例 1. 目的のキーワードリストを含むファイルを作成し ( 例 : list.txt) 2. 該当箇所を変更し R Console 画面上でコピペ 一連の作業手順を記述したスクリプトを 1 つのファイルとして保存することをお勧め list.txt ファイル作成時に membrane と打った後に改行を入れた場合と入れない場合の挙動の違いも見ておくとよい 17

18 ありがちなミス 1 作業ディレクトリの変更を忘れている 18
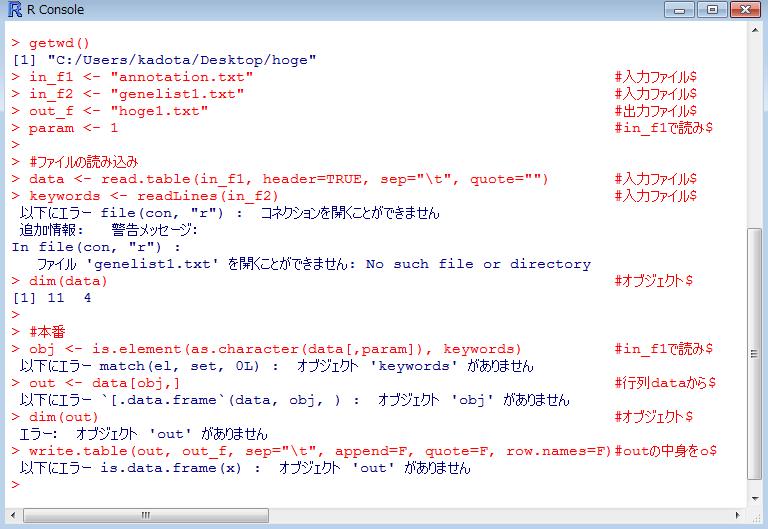

19 ありがちなミス 2 必要な入力ファイルが作業ディレクトリ中に存在しない 19

20 ありがちなミス 3 出力予定のファイル名と同じものを別のプログラムで開いているため最後の write.table 関数のところでエラーが出る 20

21 ありがちなミス 4 実行スクリプトをコピーする際 最後の行のところで改行を含ませずに R Console 画面上でペーストしたため 最後のコマンドが実行されない ( 出力ファイルが生成されない ) 21
22 参考 読み込み 1. 目的のキーワードリストを含むファイルを作成し 例 :list.txt 2. 該当箇所を変更し R コンソール画面上でコピペ in_f1 で指定したファイルを読み込め 2 読み込むファイルの最初の行はヘッダー部分です 3 ファイルの区切り文字はタブです 22

23 行列 data 参考 入力ファイルの中身を正しく読み込めていることがわかる 23

24 参考 行列 data オブジェクト data の行数と列数は 11 と 4 webpage 中の表記が灰色なのは 特にやらなくてもいいコマンドだから 24
![行列の要素へのアクセス 参考 data[ 行, 列 ]](/docs-images/104/162319746/images/25-0.jpg "param には 1 という数値が代入されていたから")
25 行列の要素へのアクセス 参考 data[ 行, 列 ] param には 1 という数値が代入されていたから 25

26 やりたかったことをおさらい 参考 genelist1.txt 論理値ベクトル obj を用いて TRUE の要素に対応する行を抽出している 26



27 R-Tips でお勉強 参考 ときどき R-Tips に立ち返るといいと思います 27

28 論理値ベクトルを理解 genelist1.txt 参考 28

29 論理値ベクトルを理解 genelist1.txt 参考 疑問に思ったら 自分の理解できるところから試す 29
30 Contents(R で...) ゲノム解析 アノテーションファイルを読み込んで目的のキーワードを含む行のみ抽出 multi-fasta ファイルを自在に解析 配列長分布 GC 含量 フィルタリング 部分配列の切り出しなど 連続塩基の出現頻度 (CpG) 解析 ゲノム配列取得など トランスクリプトーム解析 研究目的別留意点 : サンプル内とサンプル間の違い マッピング カウント情報取得 データを眺める : クラスタリングや M-A plot 理想的な実験デザイン なぜ x 倍発現変動という議論がだめなんですか? モデルとか分布って 自分の解析結果にどういう影響を与えているの? 多重比較問題 :FDR って何? 30



31 multi-fasta ファイルからの各種情報抽出 multi-fasta って何? 31

32 multi-fasta ファイルからの各種情報抽出 R で multi-fasta ファイルを読み込んで自在に解析できます 32


33 コピー (CTRL+ALT+ 左クリック )& ペースト 一連のコマンド群をコピーして 2R Console 画面上でペースト hoge フォルダに hoge1.txt が作成されているはず 33

34 結果ファイルを眺めて動作確認 入力 : hoge4.fa 出力 : hoge1.txt 34
35 参考 N50 アセンブルがどれだけうまくいっているかを表す指標の一つ 長いコンティグから足していって Total_length の 50% に達したときのコンティグの長さ contig_2 (103 bp) Total_length / 2 (120.5 bp) contig_3 (65 bp) contig_4 (49 bp) contig_1 (24 bp) Total_length (241 bp) average だと外れ値の影響を受けやすく median だと短いコンティグが多くを占める場合に不都合らしい... 35


36 情報抽出手順の一部 参考 width 関数を使えば配列長情報を取り出せるようだ 36

37 情報抽出手順の一部 参考 50 bp 以上のコンティグからなるサブセットの抽出ができそうだ! 37


38 コードの中身が分かると応用範囲が拡大 参考 指定した配列長以下のものを抽出したいときは <= とすればよい 38


39 参考 入力 :sample1.fasta >kadota AGTGACGGTCTT 出力 :hoge1.txt >kadota TGACGGT 39


40 参考 入力 :sample1.fasta >kadota AGTGACGGTCTT 出力 :hoge1.txt >kadota TGACGGT subseq 関数は 塩基配列, start, end という形式がデフォルトのようだ 40

41 関数の使用法について 参考? 関数名で使用法を記したウェブページが開く ページの下のほうに 大抵の場合使用例が掲載されている 使用法既知の関数のマニュアルをいくつか読んで慣れておく 41

42 参考 原因既知状態で意図的にエラーを出す 入力 :sample1.fasta >kadota AGTGACGGTCTT 出力 :hoge1.txt >kadota TGACGGT マニュアルの使用例をいくつか試して ステップアップ 42

43 参考 入力 1: hoge4.fa 入力 2: list_sub2.txt 出力 :hog4.txt >contig_4 CGTGCTGATT >contig_2 CTGCCT 43


44 参考 配列ごとの GC 含量を計算したいとき 44
45 Contents(R で...) ゲノム解析 アノテーションファイルを読み込んで目的のキーワードを含む行のみ抽出 multi-fasta ファイルを自在に解析 配列長分布 GC 含量 フィルタリング 部分配列の切り出しなど 連続塩基の出現頻度 (CpG) 解析 ゲノム配列取得など トランスクリプトーム解析 研究目的別留意点 : サンプル内とサンプル間の違い マッピング カウント情報取得 データを眺める : クラスタリングや M-A plot 理想的な実験デザイン なぜ x 倍発現変動という議論がだめなんですか? モデルとか分布って 自分の解析結果にどういう影響を与えているの? 多重比較問題 :FDR って何? 45
 よりもかなり低い 期待値 ゲノム中の GC 含量を考慮した場合 : 約 41%(A:0.295, C:0.205, G: 0.205, T:0.295) なので 0.205 0.205= 4.2% ゲノム中の GC 含量を考慮しない場合 : 50%(A:0.25, C:0.25, G: 0.25, T:0.")
46 Lander et al., Nature, 409: , 2001 ヒトゲノム中の CpG 出現確率は低い 全部で 16 通りの 2 連続塩基の出現頻度分布を調べると CG となる確率の実測値 (0.986%) は期待値 (4.2%) よりもかなり低い 期待値 ゲノム中の GC 含量を考慮した場合 : 約 41%(A:0.295, C:0.205, G: 0.205, T:0.295) なので = 4.2% ゲノム中の GC 含量を考慮しない場合 : 50%(A:0.25, C:0.25, G: 0.25, T:0.25) なので = 6.25% R で調べることができます 46


47 2 連続塩基の出現頻度 : 基本形 出力 :hoge1.txt 47
48 2 連続塩基の出現確率 : 基本形 出力 :hoge2.txt 48



49 2 連続塩基の出現確率 : ヒトゲノム 出力 :hoge5.txt 49

50 s 様々な生物種のゲノム配列が R のパッケージとして提供されています 50
51 ヒトゲノム (BSgenome.Hsapiens.UCSC.hg19) の 2 連続塩基出現頻度計算ができたのは このパッケージをインストール済みだからです 51
52 参考 もしゼブラフィッシュ (BSgenome.Drerio.UCSC.danRer7) ゲノムがインストールされていなければ... 52
")
53 参考 available.genomes() でリストアップされているパッケージ名を指定可能です 53

54 multi-fasta ファイルとして保存したい場合 フリーズするので 得られた hoge5.txt をテキストエディタで開くことはやめましょう 内部的には 1writeXStringSet 関数を用いて 2fasta オブジェクトを 3out_f で指定したファイル名で 4FASTA 形式で 51 行あたりの文字数を 50 文字にして保存しています

55 fasta オブジェクトの中身 R のほうが全体像の俯瞰が容易ですよね 55

56 fasta オブジェクトの中身 最初の 24 個分を表示させたい場合 56
57 2 連続塩基の出現確率 : ヒトゲノムファイル ヒトゲノムファイル hoge5.txt を入力ファイルとして与えるやり方でもよい 57

58 パッケージって何? 参考 R を再起動した状態で? 関数名と打ち込んでも 使用法を記したウェブページが開かずにエラーが出ることがあります 58

59 パッケージって何? 参考 Biostrings というパッケージを library 関数を用いて読み込むことによって dinucleotidefrequency のような Biostrings が提供する関数群を利用できるんです 59


60 参考 パッケージを個別にインストールする場合 使い方の解説記事は PDF のところをクリック 60
")
61 参考 Biostrings 中の関数を使いこなせると 他の自然言語処理系プログラミング言語 (perl や ruby) を改めて勉強しなくても必要な解析の大部分が可能です 61
62 Contents(R で...) ゲノム解析 アノテーションファイルを読み込んで目的のキーワードを含む行のみ抽出 multi-fasta ファイルを自在に解析 配列長分布 GC 含量 フィルタリング 部分配列の切り出しなど 連続塩基の出現頻度 (CpG) 解析 ゲノム配列取得など トランスクリプトーム解析 研究目的別留意点 : サンプル内とサンプル間の違い マッピング カウント情報取得 データを眺める : クラスタリングや M-A plot 理想的な実験デザイン なぜ x 倍発現変動という議論がだめなんですか? モデルとか分布って 自分の解析結果にどういう影響を与えているの? 多重比較問題 :FDR って何? 62
63 参考 トランスクリプトームとは ある状態のあるサンプルのあるゲノムの領域 ヒト 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 遺伝子全体 ( ゲノム ) どの染色体上のどの領域にどの遺伝子があるかは調べる個体が同じなら 目だろうが心臓だろうが不変 AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA 転写物全体 ( トランスクリプトーム ) 遺伝子 1 は沢山転写されている ( 発現している ) 遺伝子 4 はごくわずかしか転写されてない 63
64 トランスクリプトームとは ある状態のあるサンプルのあるゲノムの領域 光刺激 ヒト 参考 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 遺伝子全体 ( ゲノム ) どの染色体上のどの領域にどの遺伝子があるかは調べる個体が同じなら 目だろうが心臓だろうが不変 AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA AAAAAAA 転写物全体 ( トランスクリプトーム ) 遺伝子 2 は光刺激に応答して発現亢進 遺伝子 4 も光刺激に応答して発現亢進 64
 の目のトランスクリプトーム 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 マイクロアレイ")
65 トランスクリプトーム情報を得る手段 参考 光刺激前 (T1) の目のトランスクリプトーム 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 これがいわゆる遺伝子発現行列 光刺激後 (T2) の目のトランスクリプトーム 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 マイクロアレイ RNA-Seq 65
66 トランスクリプトーム取得 次世代シーケンサー :Illumina 社の場合 光刺激前 (T1) の目のトランスクリプトーム 配列決定 参考 ペアードエンド法断片配列の両末端が数百塩基以内の対の二種類の配列が得られる 数百塩基程度に断片化 シングルエンド法 約 塩基 二種類のアダプター配列を両末端に付加 シングルエンド法の場合 アダプター 1 アダプター 2 数百塩基程度 66

)%%%++)(%%%%).")
67 参考 FASTA 形式と FASTQ 形式 FASTA 形式 1 行目 : > ではじまる一行の description 行 2 行目 : 配列情報 >SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT FASTQ 形式 1 行目 ではじまる 1 行の description 行 2 行目 : 配列情報 3 行目 : + からはじまる 1 行 ( の description 行 ) 4 行目 : GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT +!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC
68 トランスクリプトーム解析の目的は様々 トランスクリプトーム配列取得 ゲノム配列既知の場合 Cufflinks などを用いて遺伝子構造推定 ( アノテーション ) ゲノム配列未知の場合 Trinity などのトランスクリプトーム用アセンブラを実行 遺伝子または isoform ごとの発現量の正確な推定 RSEM などを利用して発現量情報を得る ある特定のサンプル内での遺伝子間の発現量の大小関係を知りたい Length bias や GC bias などの各種補正がポイント 比較するサンプル間で発現変動している遺伝子または isoform の同定 TCC パッケージなどを利用して発現変動遺伝子 (DEG) を得る Sequence depth やサンプル間で発現している遺伝子の composition bias の補正がポイント (GO 解析など )DEG 結果を用いる多くの下流解析結果に影響を及ぼす 68

69 マップされたリード数 = 発現量ではないが 基本的なマッピングプログラム (bowtie など ) を用いた場合 リファレンス配列 : ゲノム count G G1 サンプルの RNA-Seq データ mapping 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 リファレンス配列 : トランスクリプトーム count G 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 マップされたリード数のカウント情報は 発現量推定の基本情報です 69
70 研究目的別留意点 : 遺伝子間比較 発現量補正の基本形 : RPK (Reads per kilobase) RPM (Reads per million) RPKM (Reads per kilobase per million) 定数カウント数 配列長 総リード数 同一サンプル内での異なる遺伝子間の発現レベル比較の場合 配列長由来 bias: 長いほど沢山 sequence される RPKM や FPKM などの配列長を考慮して正規化されたデータで解析 GC 含量由来 bias: カウント数の分布が GC 含量依存的である Risso et al., BMC Bioinformatics, 12: 480, 2011 Benjamini and Speed, Nucleic Acids Res., 40: e72, 2012 総リード数 ( ライブラリサイズ or sequence depth) 補正は不必要理由 : 遺伝子間の発現レベルの大小関係は定数倍しても不変 70
71 研究目的別留意点 : サンプル間比較 発現量補正の基本形 : RPK (Reads per kilobase) RPM (Reads per million) RPKM (Reads per kilobase per million) 異なるサンプル間での同一遺伝子間の発現レベル比較の場合 総リード数の違い : 総リード数が x 倍違うと全体的に x 倍変動 RPM 正規化で全体を揃えることは基本 定数カウント数 配列長 総リード数 組成の違い : サンプル特異的高発現遺伝子の存在で比較困難に TMM 正規化法 (Robinson and Oshlack, Genome Biol., 11: R25, 2010) TbT 正規化法 (Kadota et al., Algorithms Mol. Biol., 7: 5, 2012) Length bias や GC bias 補正は少なくとも理論上は不必要理由 : 同一遺伝子に対して掛かる係数はサンプル間で同じ 71
72 配列長の補正 Mortazavi et al., Nat. Methods, 5: , 2008 参考 配列長が長い遺伝子ほど沢山 sequence される それらの遺伝子上にマップされる生のリード数が増加傾向 配列長が長い遺伝子ほど発現レベルが高い傾向になる 発現レベルが同じで長さの異なる二つの mrnas AAAAAAA AAAAAAA 断片化して sequence マップされたリード数をカウント AAAAAAA AAAAAAA 1 つのサンプル内で異なる遺伝子間の発現レベルの大小関係を配列長を考慮せずに比較することはできない 72
73 Garber et al., Nat. Methods, 8: , 2011 の Fig. 3a 配列長を考慮した発現量推定のイメージ 参考 gene1: 3 exons (middle length), 14 reads mapped (low coverage) gene2: 3 exons (middle length), 56 reads mapped (high coverage) gene3: 2 exons (short length), 12 reads mapped (middle coverage) gene4: 2 exons (long length), 31 reads mapped (middle coverage) マップされたリード分布生リードカウント結果補正度の発現量 Garber et al., Nat. Methods, 8: , 2011 の Fig. 3a 長さが同じならリード数の多い方が発現量高い (gene 1 vs. 2) 長いほどマップされるリード数が多くなる効果を補正する必要がある (gene 3 vs. 4) 1 つのサンプル内で転写物または遺伝子間の発現レベルの大小を比較したい場合には配列長を考慮すべきである 73

 と等価 Reads Per Kilobase")
74 配列長の補正 前提条件 : 配列長が既知 補正の基本戦略 : 配列長で割る Mortazavi et al., Nat. Methods, 5: , 2008 参考 AAAAAAA AAAAAAA 1 / 配列長 を掛ける場合 塩基あたりの平均のリード数 を計算しているのと等価 1000 / 配列長 を掛ける場合 その遺伝子の配列長が1000bpだったときのリード数(or カウント数 ) と等価 Reads Per Kilobase (RPK) Counts Per Kilobase (CPK) 74
 になるような正規化係数を掛ける例 :sample1 の正規化係数 = 100 /")
75 マイクロアレイデータの正規化 参考 各サンプルから測定されたシグナル強度の和は一定 アレイ上の遺伝子数が少ない場合は非現実的だが 数千 ~ 数万種類の遺伝子が搭載されているので妥当という思想 グローバル正規化 背景 : サンプルごとにシグナル強度の総和は異なる対策 : 総和が任意の値 ( 例では 100) になるような正規化係数を掛ける例 :sample1 の正規化係数 = 100 /
 正規化後の総リード数が 100 万 (one million) になるように補正例 :T1 の正規化係数 = 1000000 /")
76 RNA-Seq データの正規化の一部 発現している RNA 量の総和はサンプル間で一定 参考 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 RPM 正規化 Reads Per Million mapped reads(rpm) 正規化後の総リード数が 100 万 (one million) になるように補正例 :T1 の正規化係数 = / 67 76
 per")

77 RPKM Mortazavi et al., Nat. Methods, 5: , 2008 参考 Reads per kilobase (of exon) per million (mapped reads) 配列長が 1000 bp だったとき かつ総リード数が 100 万だったときのカウント数 RPKM 1,000 1,000,000 カウント数 配列長総リード数 1,000,000,000 カウント数 配列長 総リード数 sample_length_count.txt hoge1.txt 総リード数 =
78 少ない カウント数 多い GC bias 補正の必要性 Risso et al., BMC Bioinformatics, 12: 480, 2011 の Fig.1 参考 GC 含量が多い遺伝子や少ない遺伝子上にマップされたリードカウント数は GC 含量が中程度の遺伝子に比べて少ない傾向にある 少ない 多い 78
79 GC bias 補正の必要性 Risso et al., BMC Bioinformatics, 12: 480, 2011 の Fig.1 参考 Quantile 正規化 パッケージ中のサンプルファイルを解析してみると 確かに GC bias が緩和されていることがわかる 79
80 Contents(R で...) ゲノム解析 アノテーションファイルを読み込んで目的のキーワードを含む行のみ抽出 multi-fasta ファイルを自在に解析 配列長分布 GC 含量 フィルタリング 部分配列の切り出しなど 連続塩基の出現頻度 (CpG) 解析 ゲノム配列取得など トランスクリプトーム解析 研究目的別留意点 : サンプル内とサンプル間の違い マッピング カウント情報取得 データを眺める : クラスタリングや M-A plot 理想的な実験デザイン なぜ x 倍発現変動という議論がだめなんですか? モデルとか分布って 自分の解析結果にどういう影響を与えているの? 多重比較問題 :FDR って何? 80
81 今は Linux コマンド抜きで一通り解析可能 SRAdb (Zhu ら, BMC Bioinformatics, 14: 19, 2013) 公共 DB からの RNA-seq データ (FASTQ ファイル ) 取得 QuasR (Lerch ら, unpublished) リファレンス配列 ( ゲノム or トランスクリプトーム ) へのマッピング Bowtie (Langmead ら, 2009) or SpliceMap (Au ら, 2010) を選択可能 出力は BAM 形式ファイル QC レポートも 遺伝子アノテーション情報をもとにカウントデータ取得 GenomicFeatures (Lawrence ら, 2013) で得られる TranscriptDb オブジェクトを利用 UCSC known genes や Ensembl genes のカウントデータなど TCC (Sun ら, BMC Bioinformatics, 14: 219, 2013) 内部的にedgeR (Robinsonら, 2010) やDESeq (Anders, 2010) などを用いて頑健な発現変動解析を実行 アセンブル以外なら Windows( の R) 上でどうにかなる時代がやってきました 81
: AGG 出力ファイル マッピングプログラムの出力 :( どのリードが )")
82 マッピング = 大量高速文字列検索 マップされる側のリファレンス配列 :hoge4.fa マップする側の RNA-seq データ ( リードと呼ばれる ): AGG 出力ファイル マッピングプログラムの出力 :( どのリードが ) リファレンス配列上のどの位置から転写されたものかという座標情報 82
83 QuasR パッケージを用いてマッピング Basic aligner の 1 つである bowtie (Langmead et al., 2009) を利用 マッピング時に多くのオプションを指定可能 -v : 許容するミスマッチ数を指定するオプション -v 0 は リードがリファレンスに完全一致するもののみレポート -v 2 は 2 塩基ミスマッチまで許容してマップされうる場所を探索 -m : 出力するリード条件を指定するオプション -m 1 は 複数個所にマップされるリードを除外して 1 か所にのみマップされたリードをレポート -m 3 は 合計 3 か所にマップされるリードまでをレポート --best --strata : 最も少ないミスマッチ数でマップされるもののみ出力する という意思表示 これをつけずに -v 2 -m 1 などと指定すると たとえ完全一致 ( ミスマッチ数 0) で 1 か所にのみマップされるリードがあったとしても どこか別の場所で 1 塩基ミスマッチでマップされる個所があれば マップされうる場所が 2 か所ということを意味し そのリードは出力されなくなる それを防ぐのが主な目的... Langmead et al., Genome Biol., 10: R25, 2009 デフォルトである程度よきに計らってくれるが... 実際の挙動を完全に把握できる状況で様々なオプションを試したい 83
84 マッピング時に用いるオプションの理解 マップされる側のリファレンス配列 :ref_genome.fa chr3 と chr5 の違いは 2 番目と 7 番目の塩基のみ 主に -m オプションの違いの把握が可能 84

85 マッピング時に用いるオプションの理解 マップされる側のリファレンス配列 :ref_genome.fa コピペで作成しています 85
86 マッピング時に用いるオプションの理解 マップする側の RNA-seq データ :sample_rnaseq1.fa 許容するミスマッチ数による違いや マップされるべき場所が完全に把握できるように リードの description 行に記述されている 86


87 マッピング時に用いるオプションの理解 マップする側の RNA-seq データ :sample_rnaseq1.fa コピペで作成しています 87


88 複数の RNA-seq サンプルを実行できるようにリストファイルとして与える 許容するミスマッチ数は 0 個 ( -v 0 ) 1 か所にマップされるリードのみ出力 ( -m 1 ) 88

89 QuasR パッケージを用いてマッピング 実行後 出力ファイルとして実際に取り扱うのは BAM 形式ファイルです 89
90 マッピング結果の出力ファイル形式 ゲノム上のどの位置にどのリードがマッピングされたか ( トランスクリプトームの場合どの転写物配列上のどの位置にどのリードがマッピングされたか ) を表すファイル形式は複数あります SAM (Sequence Alignment/Map) format SAMtools (Li et al., Bioinformatics, 25: , 2009) BAM (Binary Alignment/Map) format SAMtools (Li et al., Bioinformatics, 25: , 2009) BED (Browser Extensible Data) format... BEDtools (Quinlan et al., Bioinformatics, 26: , 2010) 実用上は BAM 形式 視覚上は BED 形式 90
91 マッピング結果の出力ファイル形式 BAM 形式ファイル BED 形式ファイル BED の最小限の情報は リード ID を含まない 91
92 マッピングオプションと結果の解釈 -m 1 --best --strata -v 0 :0 ミスマッチで 1 か所にのみマップされるリードを出力 マップされなかったのは 計 8 リード中 3 リード 92
93 マッピングオプションと結果の解釈 -m 1 --best --strata -v 0 :0 ミスマッチで 1 か所にのみマップされるリードを出力 完全一致でも複数個所にマップされるために落とされたリード 93
94 マッピングオプションと結果の解釈 -m 1 --best --strata -v 0 :0 ミスマッチで 1 か所にのみマップされるリードを出力 1 か所にのみマップされるがミスマッチのため落とされたリード 94

95 マッピング結果からのカウント情報取得 アノテーション情報を利用する場合 UCSC Genes, Ensembl Genes など様々なテーブル名を指定可能 gene, exon, promoter, junction など様々なレベルを指定可能 アノテーション情報がない場合 マップされたリードの和集合領域を同定したのち 領域ごとのリード数をカウント BEDtools (Quinlan et al., 2010) 中の mergebed プログラムを実行して和集合領域同定後 intersectbed プログラムを実行してリード数をカウントする作業に相当 領域 count 基本的なイメージ 95

96 マッピング結果からのカウント情報取得 アノテーション情報を利用する場合 UCSC Genes, Ensembl Genes など様々なテーブル名を指定可能 gene, exon, promoter, junction など様々なレベルを指定可能 アノテーション情報がない場合 マップされたリードの和集合領域を同定したのち 領域ごとのリード数をカウント BEDtools (Quinlan et al., 2010) 中の mergebed プログラムを実行して和集合領域同定後 intersectbed プログラムを実行してリード数をカウントする作業に相当 sample1 count sample2 複数サンプルの場合には領域が変わりうる 96



97 アノテーション情報がない場合 *_range.txt というカウントデータのファイルが作成される 97

98 マッピング結果からのカウント情報取得 *.bed *_range.txt カウント数はこちら 98

99 マッピング結果からのカウント情報取得 リストファイル中で指定したサンプル名がカウントデータ行列の列名となる 99
100 よく見かけるカウントデータ取得手段 basic aligner の 1 つである Bowtie を利用 最大 2 塩基ミスマッチまで許容してリファレンス配列の 1 か所とのみ一致するリード (uniquely mapped reads or unique mapper) 数をカウント Marioni et al., Genome Res., 18: , 2008 Bullard et al., BMC Bioinformatics, 11:94, 2010 Risso et al., BMC Bioinformatics, 12:480, 2011 ReCount (Frazee et al., BMC Bioinformatics, 12:449, 2011) SpliceMap (Au et al., 2010) などの splice-aware aligner だと相当時間がかかるという現実的な問題もあるのだろう 講義や講習会では到底無理 ユーザの記憶に残らない 実際に使われない
101 定量化 : 遺伝子レベル isoform レベル 全体的な流れとしては遺伝子レベル isoform レベル 例 : 新規 splice variant の発見 (Twine et al., PLoS One, 6: e16266, 2011) 遺伝子セット解析 (Gene Ontology 解析やパスウェイ解析など ) のための基本情報は遺伝子レベルの解像度 複数エクソン 遺伝子レベルの要約統計量 exon union method (Mortazavi et al., Nat. Methods, 5: , 2008) 全ての isoforms 間で用いられている exon の情報 (union: 和集合 ) を利用 exon intersection method (Bullard et al., BMC Bioinformatics, 11: 94, 2010) 複数 isoforms 間で共通して用いられている exon の情報のみ (intersection: 積集合 ) を利用 count 情報を得る際に どの exon の情報を用いるか? 101
102 遺伝子のカウント数の定義 算出された生リードカウント結果 Garber et al., Nat. Methods, 8: , 2011 の Fig. 3c exon union method( 和集合 ) の場合 :20 reads Exon intersection method( 積集合 ) の場合 :11 reads Garber et al., Nat. Methods, 8: , 2011 の Fig. 3c 様々な思想があり 当然その後の解析結果に影響を及ぼします 102
103 Contents(R で...) ゲノム解析 アノテーションファイルを読み込んで目的のキーワードを含む行のみ抽出 multi-fasta ファイルを自在に解析 配列長分布 GC 含量 フィルタリング 部分配列の切り出しなど 連続塩基の出現頻度 (CpG) 解析 ゲノム配列取得など トランスクリプトーム解析 研究目的別留意点 : サンプル内とサンプル間の違い マッピング カウント情報取得 データを眺める : クラスタリングや M-A plot 理想的な実験デザイン なぜ x 倍発現変動という議論がだめなんですか? モデルとか分布って 自分の解析結果にどういう影響を与えているの? 多重比較問題 :FDR って何? 103


104 サンプル間クラスタリング 出力 :hoge1.png ゼロカウントを含む低発現データのフィルタリングは重要です 104
105 G2 で高発現 non-deg DEG DEG サンプル間クラスタリング data_hypodata_3vs3.txt(2 群間比較用 ) G1 群 :3サンプル G2 群 :3サンプル全部で10,000 行 6 列 最初の2,000 行分が発現変動遺伝子 (DEG) 出力 :hoge1.png non-deg G1 で高発現 G1:3 反復 G2:3 反復 DEG が多く存在するほど群間で明瞭なクラスターに分かれる傾向 クラスタリング結果から DEG の有無をある程度把握可能です 105

106 サンプル間クラスタリング 出力 :hoge2.png DEG が存在しないデータの典型的なクラスタリング結果です 106
107 M=log 2 (G2)-log 2 (G1) M-A plot Dudoit et al., Stat. Sinica, 12: , 群間比較用 横軸が全体的な発現レベル 縦軸がlog 比からなるプロット 名前の由来は おそらく対数の世界での縦軸が引き算 (Minus) 横軸が平均(Average) G1 群 < G2 群 G2 群で高発現 G1 群 = G2 群 G1 群 > G2 群 G1 群で高発現 A=(log 2 (G2)+log 2 (G1))/2 低発現 全体的に 高発現 DEG が存在しないデータの M-A plot を眺めることで 縦軸の閾値のみに相当する倍率変化を用いた DEG 同定の危険性が分かります 107

108 M-A plot RPM 補正後の non-deg データを用いて M-A plot を描画します 108


109 M-A plot ここまでは 100 万に揃えるという総リード数補正結果の確認 109


110 M-A plot RPM 補正後のデータを用いて M-A plot 110
111 M-A plot 作成手順 non-deg のデータのみで RPM 正規化したデータ 各群の平均値 log 2 変換 log 2 (G2/G1) log 比 平均発現量 任意の遺伝子 gene_2002 をハイライトさせたいときは? 111

112 M-A plot gene_2002 のような G1 群で高発現遺伝子の M 値はマイナスの位置にプロットされます 112
")
113 M-A plot G2 群で 4 倍以上高発現という条件 (499 個 ) も可能です 113
")
114 M-A plot A 値が 10 より大きいという条件 (131 個 ) も可能です 114
115 M-A plot 作成手順 non-deg のデータのみで RPM 正規化したデータ 各群の平均値 log 2 変換 log 2 (G2/G1) log 比 平均発現量 どちらか一方の群で平均値がゼロカウントになる遺伝子は通常の M-A plot 上にはプロットできません 115

116 M-A plot 作成手順 non-deg のデータのみで RPM 正規化したデータ 各群の平均値 log 2 変換 参考 どちらか一方の群で平均値がゼロカウントになる遺伝子は通常の M-A plot 上にはプロットできません 116

117 M-A plot 参考 TCC を含む RNA-seq 用パッケージはプロットできます 出力 :hoge9.png 117
118 Sun et al., BMC Bioinformatics, 14: 219, 2013 参考 M-A plot:tcc パッケージの 0 カウント対策 出力 :hoge9.png 1 各群について ゼロでない平均発現量の最小値を取得 20 だったところをその値で置換 3M 値を再計算 4M-A plot の左側に 再計算して得られた M 値をプロット 118
119 Contents(R で...) ゲノム解析 アノテーションファイルを読み込んで目的のキーワードを含む行のみ抽出 multi-fasta ファイルを自在に解析 配列長分布 GC 含量 フィルタリング 部分配列の切り出しなど 連続塩基の出現頻度 (CpG) 解析 ゲノム配列取得など トランスクリプトーム解析 研究目的別留意点 : サンプル内とサンプル間の違い マッピング カウント情報取得 データを眺める : クラスタリングや M-A plot 理想的な実験デザイン なぜ x 倍発現変動という議論がだめなんですか? モデルとか分布って 自分の解析結果にどういう影響を与えているの? 多重比較問題 :FDR って何? 119
120 理想的な実験デザイン 腎臓 vs. 肝臓のような G1 群 vs. G2 群の比較の場合 生のリードカウントのデータ ( 基本的には整数値 ) Biological replicates のデータ生物学的なばらつき ( 個体間の違い ) を考慮すべし G1_rep1: ある生物の腎臓 G1_rep2: 同じ生物種の別個体の腎臓 G1_rep3: 同じ生物種のさらに別個体の腎臓 G2_rep1: ある生物の肝臓 G2_rep2: 同じ生物種の別個体の肝臓 120
121 18,110 genes 2 群間比較 :technical replicates データ data_marioni.txt ( ヒトのデータ ) Marioni et al., Genome Res., 18: , 2008 kidney( 腎臓 ) liver( 肝臓 ) Technical replicates のデータレーン間の違いなどサンプル内の技術的なばらつきの度合いを調べるための同一個体由来データ このようなデータで 2 群間比較し 発現変動遺伝子がどの程度あるかといった数に関する議論はほぼ無意味 理由 :Biological variation > Technical variation 得られた結果はその個体内のみで成立するものであり 同じ生物種の別個体においても同様な事象が観測されるわけではない 121
122 18,110 genes 2 群間比較 :technical replicates データ data_marioni.txt ( ヒトのデータ ) Marioni et al., Genome Res., 18: , 2008 kidney( 腎臓 ) liver( 肝臓 ) G1 群 G2 群 発現変動遺伝子 (DEG) がないデータで 2 群間比較をしてみると 122
123 M-A plot 左から (2+2) 列分のサブセットを抽出したうえで DEG 同定を行っています 123

124 M-A plot hoge5_fdr.png 統計的手法の FDR < 0.05 を満たす DEG 検出結果は 0 個 124


125 hoge5_fc.png 2 倍以上発現変動しているものを DEG とみなすと 18,110 遺伝子中 1,402 個も!! 125
126 Contents(R で...) ゲノム解析 アノテーションファイルを読み込んで目的のキーワードを含む行のみ抽出 multi-fasta ファイルを自在に解析 配列長分布 GC 含量 フィルタリング 部分配列の切り出しなど 連続塩基の出現頻度 (CpG) 解析 ゲノム配列取得など トランスクリプトーム解析 研究目的別留意点 : サンプル内とサンプル間の違い マッピング カウント情報取得 データを眺める : クラスタリングや M-A plot 理想的な実験デザイン なぜ x 倍発現変動という議論がだめなんですか? モデルとか分布って 自分の解析結果にどういう影響を与えているの? 多重比較問題 :FDR って何? 126
127 18,110 genes 2 群間比較 :technical replicates データ data_marioni.txt ( ヒトのデータ ) Marioni et al., Genome Res., 18: , 2008 kidney( 腎臓 ) liver( 肝臓 ) G1 群 通常の 2 群間比較をしてみると G2 群 127

128 M-A plot サブセットの抽出を行わずに DEG 同定を行っています 128


129 hoge4_fdr.png 統計的手法の FDR < 0.05 を満たす DEG 検出結果は全 18,110 個中 10,831 個 129



130 hoge4_fc.png 2 倍以上発現変動しているものを DEG とみなすと 18,110 個中 7,807 個も!! 130
131 18,110 genes 2 群間比較 :technical replicates データ data_marioni.txt ( ヒトのデータ ) TCC (Sun et al., BMC Bioinformatics, 14: 219, 2013) hoge5_fdr.png kidney( 腎臓 ) liver( 肝臓 ) この分布は同一群内のばらつきの程度を表している この分布のど真ん中に位置する遺伝子の p 値 = 1 G1 群 G2 群 131
132 18,110 genes 2 群間比較 :technical replicates データ data_marioni.txt ( ヒトのデータ ) TCC (Sun et al., BMC Bioinformatics, 14: 219, 2013) hoge4_fdr.png kidney( 腎臓 ) liver( 肝臓 ) 同一群内のばらつきの範囲内は正しく non-deg それ以外の位置が DEG と判定されていることがわかる G1 群 G2 群 132
133 モデル構築 同一群に属する反復実験データのばらつきの程度を把握すること 平均 - 分散 (MEAN-VARIANCE) プロットがよく用いられる M-A plotの場合は 縦軸のM 値 (log 比 ) がばらつきの指標に相当するが 無数の組合せが存在する 同一群 G1 群 G2 群 G1 群 G2 群 G1 群 G2 群 分散!! ばらつきの程度をより直接的に表現する指標は分散 133


134 平均 - 分散プロット ゲノム解析 アノテーションフ 実行してみましょう 134
135 平均 - 分散プロット y = x の直線上にプロットされている 入力データ (G1 群 ) 総リード数補正後のデータ 135
136 平均 - 分散プロット Technical replicates のデータは : VARIANCE はその MEAN で説明可能である (VARIANCE MEAN) ポアソン分布に従う ポアソンモデルが適用可能 hoge1_g1.png hoge1_g2.png hoge1_all.png adjusted R-squared: adjusted R-squared:
137 18,110 genes 平均 - 分散プロット :non-deg 分布を把握 kidney( 腎臓 ) liver( 肝臓 ) hoge1_g1.png hoge1_g2.png G1 群 G2 群 (G1 群 + G2 群 ) の平均 - 分散プロットを眺めると
")
138 平均 - 分散プロット hoge4.png 同一群内のばらつきの範囲 (non-deg 分布 ) 外に多数の遺伝子が存在 138

139 平均 - 分散プロット hoge5.png 一般に DEG は non-deg に比べ (G1 群 + G2 群 ) の分散が大きいので妥当 139
140 26,221 genes 2 群間比較 :biological replicates データ data_arab.txt ( 植物のデータ ) Cumbie et al., PLoS ONE, 6: e25279, 2011 G1 群 G2 群 オリジナルは AT4G32850 が重複して存在していたため 19,520 行目のデータを予め除去している 発現変動遺伝子 (DEG) がないデータで 2 群間比較をしてみると 140

")

141 M-A plot 計 6 列からなるデータから (1, 3) 列のサブセットを抽出したうえで DEG 同定を行っています 141

142 M-A plot hoge3_fdr.png 統計的手法の FDR < 0.05 を満たす DEG 検出結果は全 26,221 個中 0 個 142


143 hoge3_fc.png 2 倍以上発現変動しているものを DEG とみなすと 26,221 遺伝子中 6,641 個も!! 143
 mock 群 hrcc 群統計的手法倍率変化 G1 群 G2 群 統計的手法のほうが non-deg を DEG と判定するミス (false positives) が圧倒的に少ない")
144 性能評価 : 統計的手法 vs. 倍率変化 data_marioni.txt (technical replicates) 腎臓 (Kidney) 群 統計的手法 肝臓 (Liver) 群 倍率変化 G1 群 G2 群 data_arab.txt (biological replicates) mock 群 hrcc 群統計的手法倍率変化 G1 群 G2 群 統計的手法のほうが non-deg を DEG と判定するミス (false positives) が圧倒的に少ない 144
145 ばらつき度 :technical vs. biological data_marioni.txt (technical replicates) 腎臓 (Kidney) 群 technical replicates 肝臓 (Liver) 群 G1 群 G2 群 data_arab.txt (biological replicates) mock 群 hrcc 群 biological replicates G1 群 G2 群 Biological replicates データのほうが同一群のばらつきが大きい 145


146 平均 - 分散プロット 実行してみましょう 146
147 Biological replicates のデータは : VARIANCE > MEAN 負の二項 (NB) 分布に従う NB モデルが適用可能 hoge4_g1.png hoge4_g2.png hoge4_all.png 147
148 26,222 genes 平均 - 分散プロットの結果の解釈 分散 (VARIANCE) は平均 (MEAN) よりも大きい傾向にある VARIANCE > MEAN (y = x の灰色直線が VARIANCE = MEAN に相当 ) VARIANCE = MEAN + φ MEAN 2 (φ > 0;φ : dispersion parameter) で表現される G1 群 G2 群 負の二項分布 (negative binomial distribution; NB 分布 ) は biological replicates データのばらつきの程度を表現する基本モデル 148
149 統計的手法とは 同一群に属する反復実験データを用いてばらつきの程度を把握 モデル構築に相当 負の二項分布 (NB) モデル :VARIANCE = MEAN + φ MEAN 2 (φ > 0) Biological replicates データ表現用 ポアソン分布モデル :VARIANCE = MEAN Technical replicatesデータ表現用 NBモデルのφ = 0の場合に相当 NBモデルの数式でポアソンモデルに対応可能 発現変動していない遺伝子でもこれだけばらつくというデータの素性を知ることが重要なんです 149
150 統計的手法とは 同一群に属する反復実験データを用いてばらつきの程度を把握 モデル構築に相当 負の二項分布 (NB) モデル :VARIANCE = MEAN + φ MEAN 2 (φ > 0) Biological replicates データ表現用 ポアソン分布モデル :VARIANCE = MEAN Technical replicates データ表現用 NB モデルの φ = 0 の場合に相当 NB モデルの数式でポアソンモデルに対応可能 数式で表現するのは 検定結果として p 値を出力したいからです どの数式を使ってどれだけうまく non-deg 分布を把握しきれるかがポイント 150
151 似非 ( エセ ) 検定 基本的な考え方 評価軸 : 平均 (MEAN) と分散 (VAR) non-deg 分布 : 同一群のばらつき ( モデルに相当 ) 検定 :(G1+G2) 群の MEAN と VAR をプロット ( ) しておき そのあとに各群のプロット ( と ) を重ね書き non-deg 分布のど真ん中に位置した :p 値 = 1 non-deg 分布から遠く離れた位置の :p 値 = 0 (G1+G2) 群 G1 群 G2 群 MEAN = 100 となる DEG をプロットしてみる 151
152 似非 ( エセ ) 検定 基本的な考え方 評価軸 : 平均 (MEAN) と分散 (VAR) non-deg 分布 : 同一群のばらつき ( モデルに相当 ) 検定 :(G1+G2) 群の MEAN と VAR をプロット ( ) しておき そのあとに各群のプロット ( と ) を重ね書き non-deg 分布のど真ん中に位置した :p 値 = 1 non-deg 分布から遠く離れた位置の :p 値 = 0 (G1+G2) 群 G1 群 G2 群 DEG1( 黄色 ) DEG2( マゼンタ ) ともに non-deg 分布の端の方に位置することがわかる 152

153 平均 - 分散プロット : 実際の DEG 同定 hoge6.png FDR < 0.05 を満たす 366 個の DEG の分散は それ以外の non-deg よりも全体的に大きいので妥当 153

154 M-A plot 通常は 平均 - 分散プロットではなく M-A plot で DEG 検出結果を示します 154

155 hoge6_fdr.png FDR < 0.05 を満たすものは 366 個 155
156 出力ファイル (hoge6.txt) の説明 入力データ p-value とその順位 M-A plot の A 値と M 値 q-value FDR 閾値判定結果 q-value < 0.05 を満たす DEG が 1 non-deg が 0 基本的には これらの結果を用います 156
157 Contents(R で...) ゲノム解析 アノテーションファイルを読み込んで目的のキーワードを含む行のみ抽出 multi-fasta ファイルを自在に解析 配列長分布 GC 含量 フィルタリング 部分配列の切り出しなど 連続塩基の出現頻度 (CpG) 解析 ゲノム配列取得など トランスクリプトーム解析 研究目的別留意点 : サンプル内とサンプル間の違い マッピング カウント情報取得 データを眺める : クラスタリングや M-A plot 理想的な実験デザイン なぜ x 倍発現変動という議論がだめなんですか? モデルとか分布って 自分の解析結果にどういう影響を与えているの? 多重比較問題 :FDR って何? 157
 5% というのが q-value < 0.")
158 多重比較問題 :FDR って何? DEG か non-deg かを判定する閾値を決める問題 有意水準 5% というのが p-value < 0.05 に相当 Benjamini and Hochberg J. Roy. Stat. Soc. B, 57: , False discovery rate (FDR) 5% というのが q-value < 0.05 に相当 発現変動ランキング結果は不変なので上位 x 個という決め打ちの場合にはこの問題とは無関係 DEG 数に関するよりよい結果を得たい場合には p-value ではなく q-value 閾値を利用しましょう 158
159 多重比較問題 :FDR って何? p-value (false positive rate; FPR) 本当は DEG ではないにもかかわらず DEG と判定してしまう確率 全遺伝子に占める non-deg の割合 ( 分母は遺伝子総数 ) 例 :10,000 個の non-deg からなる遺伝子を p-value < 0.05 で検定すると 10, = 500 個程度の non-deg を間違って DEG と判定することに相当 実際の DEG 検出結果が 900 個だった場合 :500 個は偽物で 400 個は本物と判断 実際の DEG 検出結果が 510 個だった場合 :500 個は偽物で 10 個は本物と判断 実際の DEG 検出結果が 500 個以下の場合 : 全て偽物と判断 q-value (false discovery rate: FDR) DEG と判定した中に含まれる non-deg の割合 Benjamini and Hochberg J. Roy. Stat. Soc. B, 57: , DEG 中に占める non-deg の割合 ( 分母は DEG と判定された数 ) non-deg の期待値を計算できれば p 値でも上位 x 個でも DEG と判定する手段はなんでもよい 以下は 10,000 遺伝子の検定結果での FDR 計算例 p < を満たす DEG 数が 100 個の場合 :FDR = 10, /100 = 0.1 p < 0.01 を満たす DEG 数が 400 個の場合 :FDR = 10, /400 = 0.25 p < 0.05 を満たす DEG 数が 926 個の場合 :FDR = 10, /926 =
160 18,110 genes 2 群間比較 :technical replicates データ data_marioni.txt ( ヒトのデータ ) Marioni et al., Genome Res., 18: , 2008 kidney( 腎臓 ) liver( 肝臓 ) G1 群 G2 群 発現変動遺伝子 (DEG) がないデータで 2 群間比較をしてみると 160

161 M-A plot q-value < 0.05 を満たす DEG 数は 0 個 p-value < 0.05 を満たす DEG 数は 465 個 161




162 ReCount (Frazee et al., BMC Bioinformatics, 12: 449, 2011) 他の公共カウントデータでも確認できます 評価軸 使い慣れているので 私は ReCount のデータをよく利用しています 自分でもいろいろと試してみましょう 162
")
163 東大生以外の方も受講可能です ( 平成 26 年度もやります ) 163
 グラント 基盤研究 (C)(H24-26 年度 ):")
: 非モデル生物におけるゲノム解析法の確立 ( 分担 ; 研究代表者 : 西山智明 ) 挿絵や TCC のロゴなど ( 妻の )")
164 謝辞 共同研究者清水謙多郎先生 ( 東京大学 大学院農学生命科学研究科 ) 西山智明先生 ( 金沢大学 学際科学実験センター ) 孫建強氏 ( 東京大学 大学院農学生命科学研究科 大学院生 ) グラント 基盤研究 (C)(H24-26 年度 ): シークエンスに基づく比較トランスクリプトーム解析のためのガイドライン構築 ( 代表 ) 新学術領域研究 ( 研究領域提案型 )(H22 年度 -): 非モデル生物におけるゲノム解析法の確立 ( 分担 ; 研究代表者 : 西山智明 ) 挿絵や TCC のロゴなど ( 妻の ) 門田雅世さま作 ( 有能な秘書の ) 三浦文さま作 164
ゲノム情報解析基礎 ~ Rで塩基配列解析 ~
 ネット接続できないヒトも ダブルクリックでローカルに r_seq.html を起動可能です 実習は デスクトップ上にある hoge フォルダの中身が以下の状態を想定して行います (R で ) 塩基配列解析の利用法 : GC 含量計算から発現変動解析まで東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
ネット接続できないヒトも ダブルクリックでローカルに r_seq.html を起動可能です 実習は デスクトップ上にある hoge フォルダの中身が以下の状態を想定して行います (R で ) 塩基配列解析の利用法 : GC 含量計算から発現変動解析まで東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
基本的な利用法
 (R で ) 塩基配列解析 基本的な利用法 Macintosh 版 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける 1. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール 2. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握
(R で ) 塩基配列解析 基本的な利用法 Macintosh 版 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける 1. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール 2. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握
機能ゲノム学(第6回)
") トランスクリプトーム解析の今昔 なぜマイクロアレイ? なぜRNA-Seq? 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 Contents トランスクリプトーム解析の概要 各手法の長所 短所 マイクロアレイ
トランスクリプトーム解析の今昔 なぜマイクロアレイ? なぜRNA-Seq? 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 Contents トランスクリプトーム解析の概要 各手法の長所 短所 マイクロアレイ
特論I
 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 4 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp 1 前回の課題と正答 アダプター配列除去前後の small RNA-seq データをカイコゲノムにマップし マップ率 ( マップされたリード数
講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 4 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp 1 前回の課題と正答 アダプター配列除去前後の small RNA-seq データをカイコゲノムにマップし マップ率 ( マップされたリード数
機能ゲノム学(第6回)
") RNA-Seq データ解析リテラシー 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 2009 年ごろの私 次世代シーケンサー (NGS) 解析についての認識 単に短い塩基配列が沢山あるだけでしょ 得られる配列データって
RNA-Seq データ解析リテラシー 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 2009 年ごろの私 次世代シーケンサー (NGS) 解析についての認識 単に短い塩基配列が沢山あるだけでしょ 得られる配列データって
機能ゲノム学(第6回)
") RNA-Seqデータ解析における正規化法の選択 :RPKM 値でサンプル間比較は危険?! 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 よりよい正規化法とは? その正規化法によって得られたデータを用いて発現変動の度合いでランキングしたときに
RNA-Seqデータ解析における正規化法の選択 :RPKM 値でサンプル間比較は危険?! 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 よりよい正規化法とは? その正規化法によって得られたデータを用いて発現変動の度合いでランキングしたときに
機能ゲノム学(第6回)
") R でトランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 自己紹介 2002 年 3 月 東京大学 大学院農学生命科学研究科博士課程修了 学位論文 : cdna マイクロアレイを用いた遺伝子発現解析手法の開発
R でトランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 自己紹介 2002 年 3 月 東京大学 大学院農学生命科学研究科博士課程修了 学位論文 : cdna マイクロアレイを用いた遺伝子発現解析手法の開発
基本的な利用法
 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握 ) 4. ありがちなミス のところで示しているエラーメッセージとその原因をきっちり理解
到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握 ) 4. ありがちなミス のところで示しているエラーメッセージとその原因をきっちり理解
ゲノム情報解析基礎
 講義資料 PDF が講義のページからダウンロード可能です 印刷物はありません ゲノム情報解析基礎 ~ R で塩基配列解析 ~ 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム 微生物科学イノベーション連携研究機構門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 講義予定
講義資料 PDF が講義のページからダウンロード可能です 印刷物はありません ゲノム情報解析基礎 ~ R で塩基配列解析 ~ 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム 微生物科学イノベーション連携研究機構門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 講義予定
Rでトランスクリプトーム解析
 R でトランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 自己紹介 1995 年 3 月 高知工業高等専門学校 工業化学科卒業 1997 年 3 月 東京農工大学 工学部 物質生物工学科卒業
R でトランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 自己紹介 1995 年 3 月 高知工業高等専門学校 工業化学科卒業 1997 年 3 月 東京農工大学 工学部 物質生物工学科卒業
基本的な利用法
 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握 ) 4. ありがちなミス のところで示しているエラーメッセージとその原因をきっちり理解
到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握 ) 4. ありがちなミス のところで示しているエラーメッセージとその原因をきっちり理解
特論I
 2016.02.01 版 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 3 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp Jun 25, 2014 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG
2016.02.01 版 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 3 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp Jun 25, 2014 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG
農学生命情報科学特論I
 2015.07.01 版 USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 前回 (6/23) の hoge フォルダがデスクトップに残っているかもしれないのでご注意ください 農学生命情報科学 特論 I 第 3 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
2015.07.01 版 USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 前回 (6/23) の hoge フォルダがデスクトップに残っているかもしれないのでご注意ください 農学生命情報科学 特論 I 第 3 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
NGSデータ解析入門Webセミナー
 NGS データ解析入門 Web セミナー : RNA-Seq 解析編 1 RNA-Seq データ解析の手順 遺伝子発現量測定 シークエンス マッピング サンプル間比較 機能解析など 2 CLC Genomics Workbench 使用ツール シークエンスデータ メタデータのインポート NGS data import Import Metadata クオリティチェック Create Sequencing
NGS データ解析入門 Web セミナー : RNA-Seq 解析編 1 RNA-Seq データ解析の手順 遺伝子発現量測定 シークエンス マッピング サンプル間比較 機能解析など 2 CLC Genomics Workbench 使用ツール シークエンスデータ メタデータのインポート NGS data import Import Metadata クオリティチェック Create Sequencing
NGSハンズオン講習会
 205.07.27 版 配布する USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください NGS ハンズオン 講習会 :R 基礎 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ Contents(
205.07.27 版 配布する USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください NGS ハンズオン 講習会 :R 基礎 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ Contents(
Rインストール手順
 R 本体は最新のリリース版 R パッケージは 必要最小限プラスアルファ の推奨インストール手順を示します R 本体とパッケージのインストール Windows 版 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/
R 本体は最新のリリース版 R パッケージは 必要最小限プラスアルファ の推奨インストール手順を示します R 本体とパッケージのインストール Windows 版 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/
シーケンサー利用技術講習会 第10回 サンプルQC、RNAseqライブラリー作製/データ解析実習講習会
 シーケンサー利用技術講習会 第 10 回サンプル QC RNAseq ライブ ラリー作製 / データ解析実習講習会 理化学研究所ライフサイエンス技術基盤研究センターゲノムネットワーク解析支援施設田上道平 次世代シーケンサー Sequencer File Format Output(Max) Read length Illumina Hiseq2500 Fastq 600 Gb 100 bp Life
シーケンサー利用技術講習会 第 10 回サンプル QC RNAseq ライブ ラリー作製 / データ解析実習講習会 理化学研究所ライフサイエンス技術基盤研究センターゲノムネットワーク解析支援施設田上道平 次世代シーケンサー Sequencer File Format Output(Max) Read length Illumina Hiseq2500 Fastq 600 Gb 100 bp Life
NGS速習コース
 バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-3. R 各種パッケージ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-3. R 各種パッケージ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
ゲノム情報解析基礎 ~ Rで塩基配列解析 ~
 トランスクリプトーム解析の現況 ~ マイクロアレイ vs. RNA-seq~ 東京大学 大学院農学生命科学研究科 アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 スライド PDF はウェブから取得可能です 2 ステレオタイプなイメージ
トランスクリプトーム解析の現況 ~ マイクロアレイ vs. RNA-seq~ 東京大学 大学院農学生命科学研究科 アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 スライド PDF はウェブから取得可能です 2 ステレオタイプなイメージ
機能ゲノム学(第6回)
") RNAseqによる 定 量 的 解 析 とqPCR マイクロアレイなど との 比 較 東 京 大 学 大 学 院 農 学 生 命 科 学 研 究 科 アグリバイオインフォマティクス 教 育 研 究 ユニット 門 田 幸 二 (かどた こうじ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 自 己 紹 介 1995
RNAseqによる 定 量 的 解 析 とqPCR マイクロアレイなど との 比 較 東 京 大 学 大 学 院 農 学 生 命 科 学 研 究 科 アグリバイオインフォマティクス 教 育 研 究 ユニット 門 田 幸 二 (かどた こうじ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 自 己 紹 介 1995
特論I
 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 2 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG 概論 現状や展望など 講義のみ 第 2 回 (2014
講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 2 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG 概論 現状や展望など 講義のみ 第 2 回 (2014
PowerPoint プレゼンテーション
 V1 次世代シークエンサ実習 II 本講義の内容 Reseq 解析 RNA-seq 解析 公開データ取得 クオリティコントロール マッピング 変異検出 公開データ取得 クオリティコントロール マッピング 発現定量 FPKM を算出します 2 R N A - s e q とは メッセンジャー RNA(mRNA) をキャプチャして次世代シーケンサーでシーケンシングする手法 リファレンスがある生物種の場合
V1 次世代シークエンサ実習 II 本講義の内容 Reseq 解析 RNA-seq 解析 公開データ取得 クオリティコントロール マッピング 変異検出 公開データ取得 クオリティコントロール マッピング 発現定量 FPKM を算出します 2 R N A - s e q とは メッセンジャー RNA(mRNA) をキャプチャして次世代シーケンサーでシーケンシングする手法 リファレンスがある生物種の場合
IonTorrent RNA-Seq 解析概要 サーモフィッシャーサイエンティフィックライフテクノロジーズジャパンテクニカルサポート The world leader in serving science
 IonTorrent RNA-Seq 解析概要 2017-03 サーモフィッシャーサイエンティフィックライフテクノロジーズジャパンテクニカルサポート The world leader in serving science 資料概要 この資料は IonTorrent シーケンサーで RNA-Seq (WholeTranscriptome mrna ampliseqrna mirna) 解析を実施されるユーザー様向けの内容となっています
IonTorrent RNA-Seq 解析概要 2017-03 サーモフィッシャーサイエンティフィックライフテクノロジーズジャパンテクニカルサポート The world leader in serving science 資料概要 この資料は IonTorrent シーケンサーで RNA-Seq (WholeTranscriptome mrna ampliseqrna mirna) 解析を実施されるユーザー様向けの内容となっています
1. はじめに 1. はじめに 1-1. KaPPA-Average とは KaPPA-Average は KaPPA-View( でマイクロアレイデータを解析する際に便利なデータ変換ソフトウェアです 一般のマイクロアレイでは 一つのプロー
 KaPPA-Average 1.0 マニュアル 第 1.0 版 制作者 : かずさ DNA 研究所櫻井望 制作日 : 2010 年 1 月 12 日 目次 1. はじめに 2 1-1. KaPPA-Average とは 2 1-2. 動作環境 3 1-3. インストールと起動 3 2. 操作説明 4 2-1. メイン機能 - Calc. Average 4 2-1-1. データの準備 4 2-1-2.
KaPPA-Average 1.0 マニュアル 第 1.0 版 制作者 : かずさ DNA 研究所櫻井望 制作日 : 2010 年 1 月 12 日 目次 1. はじめに 2 1-1. KaPPA-Average とは 2 1-2. 動作環境 3 1-3. インストールと起動 3 2. 操作説明 4 2-1. メイン機能 - Calc. Average 4 2-1-1. データの準備 4 2-1-2.
Rでゲノム・トランスクリプトーム解析
 06.03.05 版 実習用 PC のデスクトップ上に hoge フォルダがあります この中に解析に必要な入力ファイルがあります ネットワーク不具合時は ローカル環境で html ファイルを起動して各自対応してください R で塩基配列解析 : ゲノム解析からトランスクリプトーム解析まで 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ )
06.03.05 版 実習用 PC のデスクトップ上に hoge フォルダがあります この中に解析に必要な入力ファイルがあります ネットワーク不具合時は ローカル環境で html ファイルを起動して各自対応してください R で塩基配列解析 : ゲノム解析からトランスクリプトーム解析まで 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ )
Microsoft Word - 1 color Normalization Document _Agilent version_ .doc
 color 実験の Normalization color 実験で得られた複数のアレイデータを相互比較するためには Normalization( 正規化 ) が必要です 2 つのサンプルを異なる色素でラベル化し 競合ハイブリダイゼーションさせる 2color 実験では 基本的に Dye Normalization( 色素補正 ) が適用されますが color 実験では データの特徴と実験の目的 (
color 実験の Normalization color 実験で得られた複数のアレイデータを相互比較するためには Normalization( 正規化 ) が必要です 2 つのサンプルを異なる色素でラベル化し 競合ハイブリダイゼーションさせる 2color 実験では 基本的に Dye Normalization( 色素補正 ) が適用されますが color 実験では データの特徴と実験の目的 (
機能ゲノム学
 08.05.08 版 講義資料 PDF が講義のページからダウンロード可能です 講義資料の印刷物はありません 課題用の A4 一枚はあります 第 回出席予定の持込み PC の方は 当日までに Java のインストールをしておいてください 機能ゲノム学第 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム 微生物科学イノベーション連携研究機構門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
08.05.08 版 講義資料 PDF が講義のページからダウンロード可能です 講義資料の印刷物はありません 課題用の A4 一枚はあります 第 回出席予定の持込み PC の方は 当日までに Java のインストールをしておいてください 機能ゲノム学第 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム 微生物科学イノベーション連携研究機構門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
PrimerArray® Analysis Tool Ver.2.2
 研究用 PrimerArray Analysis Tool Ver.2.2 説明書 v201801 PrimerArray Analysis Tool Ver.2.2 は PrimerArray( 製品コード PH001 ~ PH007 PH009 ~ PH015 PN001 ~ PN015) で得られたデータを解析するためのツールで コントロールサンプルと 1 種類の未知サンプル間の比較が可能です
研究用 PrimerArray Analysis Tool Ver.2.2 説明書 v201801 PrimerArray Analysis Tool Ver.2.2 は PrimerArray( 製品コード PH001 ~ PH007 PH009 ~ PH015 PN001 ~ PN015) で得られたデータを解析するためのツールで コントロールサンプルと 1 種類の未知サンプル間の比較が可能です
機能ゲノム学(第6回)
") バイオインフォマティクス次世代シーケンサー (NGS) 編 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 バイオインフォマティクス人材育成講座 スタンダードコース 2 自己紹介 1995 年 3 月 高知工業高等専門学校
バイオインフォマティクス次世代シーケンサー (NGS) 編 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 バイオインフォマティクス人材育成講座 スタンダードコース 2 自己紹介 1995 年 3 月 高知工業高等専門学校
機能ゲノム学(第6回)
") トランスクリプトーム 解析手法の開発 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス人材養成ユニット門田幸二 2008/12/08 トランスクリプトーム (transcrptome) とは 細胞中に存在する転写物全体 (transcrpt + ome) トランスクリプトーム解析技術 DNA マイクロアレイ Affymetrx GeneChp, cdna アレイ, 電気泳動に基づく方法
トランスクリプトーム 解析手法の開発 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス人材養成ユニット門田幸二 2008/12/08 トランスクリプトーム (transcrptome) とは 細胞中に存在する転写物全体 (transcrpt + ome) トランスクリプトーム解析技術 DNA マイクロアレイ Affymetrx GeneChp, cdna アレイ, 電気泳動に基づく方法
ChIP-seq
 ChIP-seq 1 ChIP-seq 解析原理 ChIP サンプルのフラグメントでは タンパク質結合部位付近にそれぞれ Forward と Reverse のリードがマップされることが予想される ChIP のサンプルでは Forward と Reverse のリードを 3 側へシフトさせ ChIP のピークを算出する コントロールサンプルでは ChIP のサンプルとは異なり 特定の場所に多くマップされないため
ChIP-seq 1 ChIP-seq 解析原理 ChIP サンプルのフラグメントでは タンパク質結合部位付近にそれぞれ Forward と Reverse のリードがマップされることが予想される ChIP のサンプルでは Forward と Reverse のリードを 3 側へシフトさせ ChIP のピークを算出する コントロールサンプルでは ChIP のサンプルとは異なり 特定の場所に多くマップされないため
ゲノム情報解析基礎
 ゲノム情報解析基礎 ~ R で塩基配列解析 ~ 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 多くのヒトが感想を述べられて 感想やコメント へのコメントいました ありがとうございます コピペではなく位置から自分が入力するのは無理そう
ゲノム情報解析基礎 ~ R で塩基配列解析 ~ 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 多くのヒトが感想を述べられて 感想やコメント へのコメントいました ありがとうございます コピペではなく位置から自分が入力するのは無理そう
GWB_RNA-Seq_
 CLC Genomics Workbench ウェブトレーニングセミナー : RNA-Seq 編 フィルジェン株式会社バイオサイエンス部 (biosupport@filgen.jp) 1 Advanced RNA-Seq プラグイン CLC Genomics Workbench 9.0 / Biomedical Genomics Workbench 3.0 以降で使用可能な無償プラグイン RNA-Seq
CLC Genomics Workbench ウェブトレーニングセミナー : RNA-Seq 編 フィルジェン株式会社バイオサイエンス部 (biosupport@filgen.jp) 1 Advanced RNA-Seq プラグイン CLC Genomics Workbench 9.0 / Biomedical Genomics Workbench 3.0 以降で使用可能な無償プラグイン RNA-Seq
GWB
 NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 CLC Genomics Workbench 使用ツール シークエンスデータのインポート NGS data import クオリティチェック QC for Sequencing Reads Trim Reads 参照ゲノム配列へのマッピング 再アライメント
NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 CLC Genomics Workbench 使用ツール シークエンスデータのインポート NGS data import クオリティチェック QC for Sequencing Reads Trim Reads 参照ゲノム配列へのマッピング 再アライメント
データ科学2.pptx
 データ科学 多重検定 2 mul%ple test False Discovery Rate 藤博幸 前回の復習 1 多くの検定を繰り返す時には 単純に個々の検定を繰り返すだけでは不十分 5% 有意水準ということは, 1000 回検定を繰り返すと, 50 回くらいは帰無仮説が正しいのに 間違って棄却されてすまうじちがあるということ ex) 1 万個の遺伝子について 正常細胞とガン細胞で それぞれの遺伝子の発現に差があるかどうかを検定
データ科学 多重検定 2 mul%ple test False Discovery Rate 藤博幸 前回の復習 1 多くの検定を繰り返す時には 単純に個々の検定を繰り返すだけでは不十分 5% 有意水準ということは, 1000 回検定を繰り返すと, 50 回くらいは帰無仮説が正しいのに 間違って棄却されてすまうじちがあるということ ex) 1 万個の遺伝子について 正常細胞とガン細胞で それぞれの遺伝子の発現に差があるかどうかを検定
NGSハンズオン講習会
 207.08.08 版 プラスアルファの内容です NGS 解析 ( 初 ~ 中級 ) ゲノムアセンブリ後の各種解析の補足資料 ( プラスアルファ ) 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム寺田朋子 門田幸二 Aug 29-30 207 Contents Gepard でドットプロット 連載第 8 回 W5-3 で最も長い sequence 同士のドットプロットを実行できなかったが
207.08.08 版 プラスアルファの内容です NGS 解析 ( 初 ~ 中級 ) ゲノムアセンブリ後の各種解析の補足資料 ( プラスアルファ ) 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム寺田朋子 門田幸二 Aug 29-30 207 Contents Gepard でドットプロット 連載第 8 回 W5-3 で最も長い sequence 同士のドットプロットを実行できなかったが
NGS速習コース
 バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-4. R Bioconductor I 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-4. R Bioconductor I 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
CLC Genomics Workbench ウェブトレーニングセミナー: 変異解析編
 CLC Genomics Workbench ウェブトレーニングセミナー : 遺伝子発現解析編 12 th Feb., 2016 フィルジェン株式会社バイオサイエンス部 biosupport@filgen.jp Feb., 2016_V2 1 遺伝子発現解析概要 本日のセミナーにおける解析の流れ及び使用するツール名 ( 図中赤枠部分 ) Case Control インポート インポート インポート
CLC Genomics Workbench ウェブトレーニングセミナー : 遺伝子発現解析編 12 th Feb., 2016 フィルジェン株式会社バイオサイエンス部 biosupport@filgen.jp Feb., 2016_V2 1 遺伝子発現解析概要 本日のセミナーにおける解析の流れ及び使用するツール名 ( 図中赤枠部分 ) Case Control インポート インポート インポート
Microsoft Word - CygwinでPython.docx
 Cygwin でプログラミング 2018/4/9 千葉 数値計算は計算プログラムを書いて行うわけですが プログラムには様々な 言語 があるので そのうちどれかを選択する必要があります プログラム言語には 人間が書いたプログラムを一度計算機用に翻訳したのち計算を実行するものと 人間が書いたプログラムを計算機が読んでそのまま実行するものとがあります ( 若干不正確な説明ですが ) 前者を システム言語
Cygwin でプログラミング 2018/4/9 千葉 数値計算は計算プログラムを書いて行うわけですが プログラムには様々な 言語 があるので そのうちどれかを選択する必要があります プログラム言語には 人間が書いたプログラムを一度計算機用に翻訳したのち計算を実行するものと 人間が書いたプログラムを計算機が読んでそのまま実行するものとがあります ( 若干不正確な説明ですが ) 前者を システム言語
RNA-seq
 RNA-seq 1 RNA-seq 解析フロー RNA-seq インポート クオリティチェック RNA-seq 発現差解析 この資料では RNA-seq からの説明となりますが インポート クオリティチェックについては サポート資料のページより内容をご確認いただけます 2 データ 発現解析用デモデータは 以下よりダウンロードいただけます ES 細胞 (ESC) と神経前駆細胞 (NPC) の発現解析を小さなデモデータで行えます
RNA-seq 1 RNA-seq 解析フロー RNA-seq インポート クオリティチェック RNA-seq 発現差解析 この資料では RNA-seq からの説明となりますが インポート クオリティチェックについては サポート資料のページより内容をご確認いただけます 2 データ 発現解析用デモデータは 以下よりダウンロードいただけます ES 細胞 (ESC) と神経前駆細胞 (NPC) の発現解析を小さなデモデータで行えます
GWB
 NGS データ解析入門 Web セミナー : De Novo シークエンス解析編 1 NGS 新規ゲノム配列解析の手順 シークエンス 遺伝子領域の検出 アセンブル データベース検索 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 前処理 コンティグ配列の作成 CLC Genomics Workbench 遺伝子領域の検出 Blast2GO PRO データベース検索
NGS データ解析入門 Web セミナー : De Novo シークエンス解析編 1 NGS 新規ゲノム配列解析の手順 シークエンス 遺伝子領域の検出 アセンブル データベース検索 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 前処理 コンティグ配列の作成 CLC Genomics Workbench 遺伝子領域の検出 Blast2GO PRO データベース検索
数量的アプローチ 年 6 月 11 日 イントロダクション データ分析をマスターする 12 のレッスン ウェブサポートページ ( 有斐閣 ) 水落研究室 R http:
 水落研究室 R http:") イントロダクション データ分析をマスターする 12 のレッスン ウェブサポートページ ( 有斐閣 ) http://yuhikaku-nibu.txt-nifty.com/blog/2017/09/22103.html 水落研究室 R http://depts.nanzan-u.ac.jp/ugrad/ps/mizuochi/r.html 1 この授業では統計ソフト R を使って分析を行います データを扱うソフトとして
イントロダクション データ分析をマスターする 12 のレッスン ウェブサポートページ ( 有斐閣 ) http://yuhikaku-nibu.txt-nifty.com/blog/2017/09/22103.html 水落研究室 R http://depts.nanzan-u.ac.jp/ugrad/ps/mizuochi/r.html 1 この授業では統計ソフト R を使って分析を行います データを扱うソフトとして
Rインストール手順
 R 本体は最新のリリース版 R パッケージは 必要最小限プラスアルファ の推奨インストール手順を示します スライドは Windows0 環境でのスクリーンショットです ウェブブラウザによって挙動が多少異なるのでご注意ください 私は Chrome を使っています R 本体とパッケージのインストール Windows 版 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二
R 本体は最新のリリース版 R パッケージは 必要最小限プラスアルファ の推奨インストール手順を示します スライドは Windows0 環境でのスクリーンショットです ウェブブラウザによって挙動が多少異なるのでご注意ください 私は Chrome を使っています R 本体とパッケージのインストール Windows 版 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二
win版8日目
 8 日目 : 項目のチェック (2) 1 日 30 分くらい,30 日で何とか R をそこそこ使えるようになるための練習帳 :Win 版 昨日は, 平均値などの基礎統計量を計算する試行錯誤へご招待しましたが (?), 今日は簡 単にやってみます そのためには,psych というパッケージが必要となりますが, パッケー ジのインストール & 読み込みの詳しい方法は, 後で説明します 以下の説明は,psych
8 日目 : 項目のチェック (2) 1 日 30 分くらい,30 日で何とか R をそこそこ使えるようになるための練習帳 :Win 版 昨日は, 平均値などの基礎統計量を計算する試行錯誤へご招待しましたが (?), 今日は簡 単にやってみます そのためには,psych というパッケージが必要となりますが, パッケー ジのインストール & 読み込みの詳しい方法は, 後で説明します 以下の説明は,psych
Qlucore_seminar_slide_180604
 シングルセル RNA-Seq のための 情報解析 フィルジェン株式会社バイオサイエンス部 (biosupport@filgen.jp) 1 シングルセル RNA-Seq シングルセル RNA-Seq のデータ解析では 通常の RNA-Seq データの解析手法に加え データセット内の各細胞の遺伝子発現プロファイルの違いを俯瞰できるような 強力な情報解析アルゴリズムと データのビジュアライズ機能を利用する必要がある
シングルセル RNA-Seq のための 情報解析 フィルジェン株式会社バイオサイエンス部 (biosupport@filgen.jp) 1 シングルセル RNA-Seq シングルセル RNA-Seq のデータ解析では 通常の RNA-Seq データの解析手法に加え データセット内の各細胞の遺伝子発現プロファイルの違いを俯瞰できるような 強力な情報解析アルゴリズムと データのビジュアライズ機能を利用する必要がある
リード・ゲノム・アノテーションインポート
 リード ゲノム アノテーションインポート 1 Location と Folder ロケーション フォルダ Genomics Workbenchではデータを以下のような階層構造で保存可能です フォルダの一番上位の階層を Location と呼び その下の階層を Folder と呼びます データの保存場所はロケーション毎に設定可能です たとえばあるデータは C ドライブに保存し あるデータは D ドライブに保存するといった事が可能です
リード ゲノム アノテーションインポート 1 Location と Folder ロケーション フォルダ Genomics Workbenchではデータを以下のような階層構造で保存可能です フォルダの一番上位の階層を Location と呼び その下の階層を Folder と呼びます データの保存場所はロケーション毎に設定可能です たとえばあるデータは C ドライブに保存し あるデータは D ドライブに保存するといった事が可能です
_unix_text_command.pptx
 Unix によるテキストファイル処理 2015/07/30 作業場所 以降の作業は 以下のディレクトリで行います ~/unix15/text/ cd コマンドを用いてディレクトリを移動し pwd コマンドを利用して カレントディレクトリが上記になっていることを確認してください 実習で使用するデータ 講習で使用するデータは以下のフォルダ内 ファイルがあることを確認してください ~/unix15/text/
Unix によるテキストファイル処理 2015/07/30 作業場所 以降の作業は 以下のディレクトリで行います ~/unix15/text/ cd コマンドを用いてディレクトリを移動し pwd コマンドを利用して カレントディレクトリが上記になっていることを確認してください 実習で使用するデータ 講習で使用するデータは以下のフォルダ内 ファイルがあることを確認してください ~/unix15/text/
Agilent 1色法 2条件比較 繰り返し実験なし
 GeneSpring GX11.0.2 ビギナーズガイド Agilent 1 色法 2 条件の比較繰り返し実験あり 適用 薬剤非投与と投与の解析 Wild type と Knock out の解析 正常細胞と病態細胞の解析 など ビギナーズガイドは 様々なマイクロアレイの実験デザインがあるなかで 実験デザインの種類ごとに適切なデータ解析の流れを 実例とともに紹介するガイドブックです ご自分の実験デザインに適合したガイドをお使いください
GeneSpring GX11.0.2 ビギナーズガイド Agilent 1 色法 2 条件の比較繰り返し実験あり 適用 薬剤非投与と投与の解析 Wild type と Knock out の解析 正常細胞と病態細胞の解析 など ビギナーズガイドは 様々なマイクロアレイの実験デザインがあるなかで 実験デザインの種類ごとに適切なデータ解析の流れを 実例とともに紹介するガイドブックです ご自分の実験デザインに適合したガイドをお使いください
GWB
 NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 参照ゲノム配列へのマッピング 再アライメント 変異検出 CLC Genomics Workbench または Biomedical Genomics Workbench
NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 参照ゲノム配列へのマッピング 再アライメント 変異検出 CLC Genomics Workbench または Biomedical Genomics Workbench
PowerPoint プレゼンテーション
 バイオインフォマティクス 講習会 V 事前準備 が完了されている方は コンテナの起動 ファイルのコピー (Windows) まで 進めておいてください メニュー 1. 環境構築の確認 2. 基本的なLinuxコマンド 3. ツールのインストール 4. NGSデータの基礎知識と前処理 5. トランスクリプトのアッセンブル 6. RNA-seqのリファレンスcDNAマッピングとFPKM 算出 7. RNA-seqのリファレンスゲノムマッピングとFPKM
バイオインフォマティクス 講習会 V 事前準備 が完了されている方は コンテナの起動 ファイルのコピー (Windows) まで 進めておいてください メニュー 1. 環境構築の確認 2. 基本的なLinuxコマンド 3. ツールのインストール 4. NGSデータの基礎知識と前処理 5. トランスクリプトのアッセンブル 6. RNA-seqのリファレンスcDNAマッピングとFPKM 算出 7. RNA-seqのリファレンスゲノムマッピングとFPKM
nagasaki_GMT2015_key09
 Workflow Variant Calling 03 長崎は遺伝研 大量遺伝情報研究室の所属です 国立遺伝学研究所 生命情報研究センター 3F 2F 欧州EBIと米国NCBIと密接に協力しながら DDBJ/EMBL/GenBank国際塩基配列データ ベースを構築しています 私たちは 塩基配列登録を支援するシステムづくり 登録データを活用するシステムづくり 高速シーケンス配列の情報解析 を行なっています
Workflow Variant Calling 03 長崎は遺伝研 大量遺伝情報研究室の所属です 国立遺伝学研究所 生命情報研究センター 3F 2F 欧州EBIと米国NCBIと密接に協力しながら DDBJ/EMBL/GenBank国際塩基配列データ ベースを構築しています 私たちは 塩基配列登録を支援するシステムづくり 登録データを活用するシステムづくり 高速シーケンス配列の情報解析 を行なっています
 GenBank クイックスタート GenBank は NLM/NCBI にて維持管理されている核酸配列データベースです また GenBank は EMBL, DDBJ と三極間で連携しながら国際核酸配列データベースを共同で構築しています これら三機関はデータを日々交換し続けており その規模は 160000 種にも及ぶ生物種の塩基配列から成り立つまでになっています この GenBank クイックスタートでは
GenBank クイックスタート GenBank は NLM/NCBI にて維持管理されている核酸配列データベースです また GenBank は EMBL, DDBJ と三極間で連携しながら国際核酸配列データベースを共同で構築しています これら三機関はデータを日々交換し続けており その規模は 160000 種にも及ぶ生物種の塩基配列から成り立つまでになっています この GenBank クイックスタートでは
> usdata01 と打ち込んでエンター キーを押すと V1 V2 V : : : : のように表示され 読み込まれていることがわかる ここで V1, V2, V3 は R が列のデータに自 動的につけた変数名である ( variable
 R による回帰分析 ( 最小二乗法 ) この資料では 1. データを読み込む 2. 最小二乗法によってパラメーターを推定する 3. データをプロットし 回帰直線を書き込む 4. いろいろなデータの読み込み方について簡単に説明する 1. データを読み込む 以下では read.table( ) 関数を使ってテキストファイル ( 拡張子が.txt のファイル ) のデー タの読み込み方を説明する 1.1
R による回帰分析 ( 最小二乗法 ) この資料では 1. データを読み込む 2. 最小二乗法によってパラメーターを推定する 3. データをプロットし 回帰直線を書き込む 4. いろいろなデータの読み込み方について簡単に説明する 1. データを読み込む 以下では read.table( ) 関数を使ってテキストファイル ( 拡張子が.txt のファイル ) のデー タの読み込み方を説明する 1.1
配付資料 自習用テキスト 解析サンプル配布ページ 2
 分子系統樹推定法 理論と応用 2009年11月6日 筑波大 院 生命環境 田辺晶史 配付資料 自習用テキスト 解析サンプル配布ページ http://www.fifthdimension.jp/documents/molphytextbook/ 2 参考書籍 分子系統学 3 参考書籍 統計的モデル選択とベイジアンMCMC 4 祖先的な形質 問題 OTU左の の色は表現型形質の状態を表している 赤と青
分子系統樹推定法 理論と応用 2009年11月6日 筑波大 院 生命環境 田辺晶史 配付資料 自習用テキスト 解析サンプル配布ページ http://www.fifthdimension.jp/documents/molphytextbook/ 2 参考書籍 分子系統学 3 参考書籍 統計的モデル選択とベイジアンMCMC 4 祖先的な形質 問題 OTU左の の色は表現型形質の状態を表している 赤と青
ビジネス統計 統計基礎とエクセル分析 正誤表
 ビジネス統計統計基礎とエクセル分析 ビジネス統計スペシャリスト エクセル分析スペシャリスト 公式テキスト正誤表と学習用データ更新履歴 平成 30 年 5 月 14 日現在 公式テキスト正誤表 頁場所誤正修正 6 知識編第 章 -3-3 最頻値の解説内容 たとえば, 表.1 のデータであれば, 最頻値は 167.5cm というたとえば, 表.1 のデータであれば, 最頻値は 165.0cm ということになります
ビジネス統計統計基礎とエクセル分析 ビジネス統計スペシャリスト エクセル分析スペシャリスト 公式テキスト正誤表と学習用データ更新履歴 平成 30 年 5 月 14 日現在 公式テキスト正誤表 頁場所誤正修正 6 知識編第 章 -3-3 最頻値の解説内容 たとえば, 表.1 のデータであれば, 最頻値は 167.5cm というたとえば, 表.1 のデータであれば, 最頻値は 165.0cm ということになります
Medical3
 Chapter 1 1.4.1 1 元配置分散分析と多重比較の実行 3つの治療法による測定値に有意な差が認められるかどうかを分散分析で調べます この例では 因子が1つだけ含まれるため1 元配置分散分析 one-way ANOVA の適用になります また 多重比較法 multiple comparison procedure を用いて 具体的のどの治療法の間に有意差が認められるかを検定します 1. 分析メニュー
Chapter 1 1.4.1 1 元配置分散分析と多重比較の実行 3つの治療法による測定値に有意な差が認められるかどうかを分散分析で調べます この例では 因子が1つだけ含まれるため1 元配置分散分析 one-way ANOVA の適用になります また 多重比較法 multiple comparison procedure を用いて 具体的のどの治療法の間に有意差が認められるかを検定します 1. 分析メニュー
多変量解析 ~ 重回帰分析 ~ 2006 年 4 月 21 日 ( 金 ) 南慶典
 南慶典") 多変量解析 ~ 重回帰分析 ~ 2006 年 4 月 21 日 ( 金 ) 南慶典 重回帰分析とは? 重回帰分析とは複数の説明変数から目的変数との関係性を予測 評価説明変数 ( 数量データ ) は目的変数を説明するのに有効であるか得られた関係性より未知のデータの妥当性を判断する これを重回帰分析という つまり どんなことをするのか? 1 最小 2 乗法により重回帰モデルを想定 2 自由度調整済寄与率を求め
多変量解析 ~ 重回帰分析 ~ 2006 年 4 月 21 日 ( 金 ) 南慶典 重回帰分析とは? 重回帰分析とは複数の説明変数から目的変数との関係性を予測 評価説明変数 ( 数量データ ) は目的変数を説明するのに有効であるか得られた関係性より未知のデータの妥当性を判断する これを重回帰分析という つまり どんなことをするのか? 1 最小 2 乗法により重回帰モデルを想定 2 自由度調整済寄与率を求め
PowerPoint Presentation
 エピジェノミクス解析編 2016/08/10 Filgen ChIP-seq (Transfactor & Histone), Bisulfite webex seminar 株式会社キアゲンアプライドアドバンストゲノミクス宮本真理, PhD 1 アジェンダ ChIP-seq 解析 Transcription Factor ChIP-seq Histone ChIP-seq Bisulfite-seq
エピジェノミクス解析編 2016/08/10 Filgen ChIP-seq (Transfactor & Histone), Bisulfite webex seminar 株式会社キアゲンアプライドアドバンストゲノミクス宮本真理, PhD 1 アジェンダ ChIP-seq 解析 Transcription Factor ChIP-seq Histone ChIP-seq Bisulfite-seq
統計的データ解析
 統計的データ解析 011 011.11.9 林田清 ( 大阪大学大学院理学研究科 ) 連続確率分布の平均値 分散 比較のため P(c ) c 分布 自由度 の ( カイ c 平均値 0, 標準偏差 1の正規分布 に従う変数 xの自乗和 c x =1 が従う分布を自由度 の分布と呼ぶ 一般に自由度の分布は f /1 c / / ( c ) {( c ) e }/ ( / ) 期待値 二乗 ) 分布 c
統計的データ解析 011 011.11.9 林田清 ( 大阪大学大学院理学研究科 ) 連続確率分布の平均値 分散 比較のため P(c ) c 分布 自由度 の ( カイ c 平均値 0, 標準偏差 1の正規分布 に従う変数 xの自乗和 c x =1 が従う分布を自由度 の分布と呼ぶ 一般に自由度の分布は f /1 c / / ( c ) {( c ) e }/ ( / ) 期待値 二乗 ) 分布 c
141025mishima
 NGS (RNAseq) »NGS Now Generation Sequencer»NGS»» 4 NGS(Next Generation Sequencer) Now Generation Sequencer http://www.youtube.com/watch?v=womkfikwlxm http://www.youtube.com/watch?v=mxkya9xcvbq http://www.youtube.com/watch?v=nhcj8ptycfc
NGS (RNAseq) »NGS Now Generation Sequencer»NGS»» 4 NGS(Next Generation Sequencer) Now Generation Sequencer http://www.youtube.com/watch?v=womkfikwlxm http://www.youtube.com/watch?v=mxkya9xcvbq http://www.youtube.com/watch?v=nhcj8ptycfc
パッケージのインストール Rには 複雑な解析を便利に行うためのパッケージが容易されています ( 世界中の研究者達が提供してくれる ) 今回は例として多重比較検定用のmultcomp パッケージをインストールしてみます ( 注意 ) 滋賀県立大学のようにプロキシ経由でインターネットに接続する環境で R
 今回は例として多重比較検定用のmultcomp パッケージをインストールしてみます ( 注意 ) 滋賀県立大学のようにプロキシ経由でインターネットに接続する環境で R") ソフトウェア R を用いた統計解析 清水顕史 R のインストール R の情報 ( 日本語 ) は RjpWikihttp://www.okada.jp.org/RWiki/?RjpWiki にまとめられています 説明に従って最新版の exe ファイルをダウンロード (http://cran.md.tsukuba.ac.jp/bin/windows/base/) し クリックしてインストールします インストール終了後
ソフトウェア R を用いた統計解析 清水顕史 R のインストール R の情報 ( 日本語 ) は RjpWikihttp://www.okada.jp.org/RWiki/?RjpWiki にまとめられています 説明に従って最新版の exe ファイルをダウンロード (http://cran.md.tsukuba.ac.jp/bin/windows/base/) し クリックしてインストールします インストール終了後
スライド 1
 第 6 章表計算 B(Excel 2003) ( 解答と解説 ) 6B-1. 表計算ソフトの操作 1 条件付き書式の設定 1. ( ア )=E ( イ )= お 条件付き書式とは セルの数値によりセルの背景に色を付けたり 文字に色を付けたり アイコンをつけたりして分類することができる機能です 本問題では 以下の手順が解答となります 1 2 ユーザー定義の表示形式 1. ( ア )=2 ( イ )=4
第 6 章表計算 B(Excel 2003) ( 解答と解説 ) 6B-1. 表計算ソフトの操作 1 条件付き書式の設定 1. ( ア )=E ( イ )= お 条件付き書式とは セルの数値によりセルの背景に色を付けたり 文字に色を付けたり アイコンをつけたりして分類することができる機能です 本問題では 以下の手順が解答となります 1 2 ユーザー定義の表示形式 1. ( ア )=2 ( イ )=4
初めてのプログラミング
 Excel の使い方 2 ~ 数式の入力 グラフの作成 ~ 0. データ処理とグラフの作成 前回は エクセルを用いた表の作成方法について学びました 今回は エクセルを用いたデータ処理方法と グラフの作成方法について学ぶことにしましょう 1. 数式の入力 1 ここでは x, y の値を入力していきます まず 前回の講義を参考に 自動補間機能を用いて x の値を入力してみましょう 補間方法としては A2,
Excel の使い方 2 ~ 数式の入力 グラフの作成 ~ 0. データ処理とグラフの作成 前回は エクセルを用いた表の作成方法について学びました 今回は エクセルを用いたデータ処理方法と グラフの作成方法について学ぶことにしましょう 1. 数式の入力 1 ここでは x, y の値を入力していきます まず 前回の講義を参考に 自動補間機能を用いて x の値を入力してみましょう 補間方法としては A2,
第4回
 Excel で度数分布表を作成 表計算ソフトの Microsoft Excel を使って 度数分布表を作成する場合 関数を使わなくても 四則演算(+ */) だけでも作成できます しかし データ数が多い場合に度数を求めたり 度数などの合計を求めるときには 関数を使えばデータを処理しやすく なります 度数分布表の作成で使用する関数 合計は SUM SUM( 合計を計算する ) 書式 :SUM( 数値数値
Excel で度数分布表を作成 表計算ソフトの Microsoft Excel を使って 度数分布表を作成する場合 関数を使わなくても 四則演算(+ */) だけでも作成できます しかし データ数が多い場合に度数を求めたり 度数などの合計を求めるときには 関数を使えばデータを処理しやすく なります 度数分布表の作成で使用する関数 合計は SUM SUM( 合計を計算する ) 書式 :SUM( 数値数値
Microsoft PowerPoint ppt
 情報科学第 07 回データ解析と統計代表値 平均 分散 度数分布表 1 本日の内容 データ解析とは 統計の基礎的な値 平均と分散 度数分布表とヒストグラム 講義のページ 第 7 回のその他の欄に 本日使用する教材があります 171025.xls というファイルがありますので ダウンロードして デスクトップに保存してください 2/45 はじめに データ解析とは この世の中には多くのデータが溢れています
情報科学第 07 回データ解析と統計代表値 平均 分散 度数分布表 1 本日の内容 データ解析とは 統計の基礎的な値 平均と分散 度数分布表とヒストグラム 講義のページ 第 7 回のその他の欄に 本日使用する教材があります 171025.xls というファイルがありますので ダウンロードして デスクトップに保存してください 2/45 はじめに データ解析とは この世の中には多くのデータが溢れています
 UCSC ゲノムブラウザチュートリアル UCSC ゲノムブラウザはゲノム解読がなされている真核生物を対象として自動アノテーションを行い その結果をデータベースとして公開している UCSC が進めているプロジェクトです NCBI MapViewer のようにゲノムベースでその上にアノテーションされている遺伝子などの情報を閲覧すると共に ホモロジー検索や必要なデータのダウンロードなどの機能を提供しています
UCSC ゲノムブラウザチュートリアル UCSC ゲノムブラウザはゲノム解読がなされている真核生物を対象として自動アノテーションを行い その結果をデータベースとして公開している UCSC が進めているプロジェクトです NCBI MapViewer のようにゲノムベースでその上にアノテーションされている遺伝子などの情報を閲覧すると共に ホモロジー検索や必要なデータのダウンロードなどの機能を提供しています
分析のステップ Step 1: Y( 目的変数 ) に対する値の順序を確認 Step 2: モデルのあてはめ を実行 適切なモデルの指定 Step 3: オプションを指定し オッズ比とその信頼区間を表示 以下 このステップに沿って JMP の操作をご説明します Step 1: Y( 目的変数 ) の
 に対する値の順序を確認 Step 2: モデルのあてはめ を実行 適切なモデルの指定 Step 3: オプションを指定し オッズ比とその信頼区間を表示 以下 このステップに沿って JMP の操作をご説明します Step 1: Y( 目的変数 ) の") JMP によるオッズ比 リスク比 ( ハザード比 ) の算出と注意点 SAS Institute Japan 株式会社 JMP ジャパン事業部 2011 年 10 月改定 1. はじめに 本文書は JMP でロジスティック回帰モデルによるオッズ比 比例ハザードモデルによるリスク比 それぞれに対する信頼区間を求める操作方法と注意点を述べたものです 本文書は JMP 7 以降のバージョンに対応しております
JMP によるオッズ比 リスク比 ( ハザード比 ) の算出と注意点 SAS Institute Japan 株式会社 JMP ジャパン事業部 2011 年 10 月改定 1. はじめに 本文書は JMP でロジスティック回帰モデルによるオッズ比 比例ハザードモデルによるリスク比 それぞれに対する信頼区間を求める操作方法と注意点を述べたものです 本文書は JMP 7 以降のバージョンに対応しております
Microsoft Word - å“Ÿåłžå¸°173.docx
 回帰分析 ( その 3) 経済情報処理 価格弾力性の推定ある商品について その購入量を w 単価を p とし それぞれの変化量を w p で表 w w すことにする この時 この商品の価格弾力性 は により定義される これ p p は p が 1 パーセント変化した場合に w が何パーセント変化するかを示したものである ここで p を 0 に近づけていった極限を考えると d ln w 1 dw dw
回帰分析 ( その 3) 経済情報処理 価格弾力性の推定ある商品について その購入量を w 単価を p とし それぞれの変化量を w p で表 w w すことにする この時 この商品の価格弾力性 は により定義される これ p p は p が 1 パーセント変化した場合に w が何パーセント変化するかを示したものである ここで p を 0 に近づけていった極限を考えると d ln w 1 dw dw
コマンド入力による操作1(ロード、プロット、画像ファイル出力等)
") コマンド入力による操作 1 ( ロード プロット 画像ファイル出力等 ) IUGONET データ解析講習会 平成 25 年 8 月 21 日 場所 : 国立極地研究所 東北大学八木学 yagi@pparc.gp.tohoku.ac.jp CUI の基本的な使い方の流れ 1. 初期化する 2. 解析したい期間 (timespan) を指定する 3. ロードプロシージャを用いてデータを読み込む 4. 読み込まれたデータを確認する
コマンド入力による操作 1 ( ロード プロット 画像ファイル出力等 ) IUGONET データ解析講習会 平成 25 年 8 月 21 日 場所 : 国立極地研究所 東北大学八木学 yagi@pparc.gp.tohoku.ac.jp CUI の基本的な使い方の流れ 1. 初期化する 2. 解析したい期間 (timespan) を指定する 3. ロードプロシージャを用いてデータを読み込む 4. 読み込まれたデータを確認する
Excelによる統計分析検定_知識編_小塚明_5_9章.indd
 第7章57766 検定と推定 サンプリングによって得られた標本から, 母集団の統計的性質に対して推測を行うことを統計的推測といいます 本章では, 推測統計の根幹をなす仮説検定と推定の基本的な考え方について説明します 前章までの知識を用いて, 具体的な分析を行います 本章以降の知識は操作編での操作に直接関連していますので, 少し聞きなれない言葉ですが, 帰無仮説 有意水準 棄却域 などの意味を理解して,
第7章57766 検定と推定 サンプリングによって得られた標本から, 母集団の統計的性質に対して推測を行うことを統計的推測といいます 本章では, 推測統計の根幹をなす仮説検定と推定の基本的な考え方について説明します 前章までの知識を用いて, 具体的な分析を行います 本章以降の知識は操作編での操作に直接関連していますので, 少し聞きなれない言葉ですが, 帰無仮説 有意水準 棄却域 などの意味を理解して,
Microsoft Word - 操作マニュアル-Excel-2.doc
 Excel プログラム開発の練習マニュアルー 1 ( 関数の学習 ) 作成 2015.01.31 修正 2015.02.04 本マニュアルでは Excel のプログラム開発を行なうに当たって まずは Excel の関数に関する学習 について記述する Ⅰ.Excel の関数に関する学習 1. 初めに Excel は単なる表計算のソフトと思っている方も多いと思います しかし Excel には 一般的に使用する
Excel プログラム開発の練習マニュアルー 1 ( 関数の学習 ) 作成 2015.01.31 修正 2015.02.04 本マニュアルでは Excel のプログラム開発を行なうに当たって まずは Excel の関数に関する学習 について記述する Ⅰ.Excel の関数に関する学習 1. 初めに Excel は単なる表計算のソフトと思っている方も多いと思います しかし Excel には 一般的に使用する
我々のビッグデータ処理の新しい産業応用 広告やゲーム レコメンだけではない 個別化医療 ( ライフサイエンス ): 精神神経系疾患 ( うつ病 総合失調症 ) の網羅的ゲノム診断法の開発 全人類のゲノム解析と個別化医療実現を目標 ゲノム育種 ( グリーンサイエンス ): ブルーベリー オオムギ イネ
: 精神神経系疾患 ( うつ病 総合失調症 ) の網羅的ゲノム診断法の開発 全人類のゲノム解析と個別化医療実現を目標 ゲノム育種 ( グリーンサイエンス ): ブルーベリー オオムギ イネ") モンテカルロ法による分子進化の分岐図作成 のための最適化法 石井一夫 1 松田朋子 2 古崎利紀 1 後藤哲雄 2 1 東京農工大学 2 茨城大学 2013 9 9 2013 1 我々のビッグデータ処理の新しい産業応用 広告やゲーム レコメンだけではない 個別化医療 ( ライフサイエンス ): 精神神経系疾患 ( うつ病 総合失調症 ) の網羅的ゲノム診断法の開発 全人類のゲノム解析と個別化医療実現を目標
モンテカルロ法による分子進化の分岐図作成 のための最適化法 石井一夫 1 松田朋子 2 古崎利紀 1 後藤哲雄 2 1 東京農工大学 2 茨城大学 2013 9 9 2013 1 我々のビッグデータ処理の新しい産業応用 広告やゲーム レコメンだけではない 個別化医療 ( ライフサイエンス ): 精神神経系疾患 ( うつ病 総合失調症 ) の網羅的ゲノム診断法の開発 全人類のゲノム解析と個別化医療実現を目標
日心TWS
 2017.09.22 (15:40~17:10) 日本心理学会第 81 回大会 TWS ベイジアンデータ解析入門 回帰分析を例に ベイジアンデータ解析 を体験してみる 広島大学大学院教育学研究科平川真 ベイジアン分析のステップ (p.24) 1) データの特定 2) モデルの定義 ( 解釈可能な ) モデルの作成 3) パラメタの事前分布の設定 4) ベイズ推論を用いて パラメタの値に確信度を再配分ベイズ推定
2017.09.22 (15:40~17:10) 日本心理学会第 81 回大会 TWS ベイジアンデータ解析入門 回帰分析を例に ベイジアンデータ解析 を体験してみる 広島大学大学院教育学研究科平川真 ベイジアン分析のステップ (p.24) 1) データの特定 2) モデルの定義 ( 解釈可能な ) モデルの作成 3) パラメタの事前分布の設定 4) ベイズ推論を用いて パラメタの値に確信度を再配分ベイズ推定
基礎統計
 基礎統計 第 11 回講義資料 6.4.2 標本平均の差の標本分布 母平均の差 標本平均の差をみれば良い ただし, 母分散に依存するため場合分けをする 1 2 3 分散が既知分散が未知であるが等しい分散が未知であり等しいとは限らない 1 母分散が既知のとき が既知 標準化変量 2 母分散が未知であり, 等しいとき 分散が未知であるが, 等しいということは分かっているとき 標準化変量 自由度 の t
基礎統計 第 11 回講義資料 6.4.2 標本平均の差の標本分布 母平均の差 標本平均の差をみれば良い ただし, 母分散に依存するため場合分けをする 1 2 3 分散が既知分散が未知であるが等しい分散が未知であり等しいとは限らない 1 母分散が既知のとき が既知 標準化変量 2 母分散が未知であり, 等しいとき 分散が未知であるが, 等しいということは分かっているとき 標準化変量 自由度 の t
Kumamoto University Center for Multimedia and Information Technologies Lab. 熊本大学アプリケーション実験 ~ 実環境における無線 LAN 受信電波強度を用いた位置推定手法の検討 ~ InKIAI 宮崎県美郷
 熊本大学アプリケーション実験 ~ 実環境における無線 LAN 受信電波強度を用いた位置推定手法の検討 ~ InKIAI プロジェクト @ 宮崎県美郷町 熊本大学副島慶人川村諒 1 実験の目的 従来 信号の受信電波強度 (RSSI:RecevedSgnal StrengthIndcator) により 対象の位置を推定する手法として 無線 LAN の AP(AccessPont) から受信する信号の減衰量をもとに位置を推定する手法が多く検討されている
熊本大学アプリケーション実験 ~ 実環境における無線 LAN 受信電波強度を用いた位置推定手法の検討 ~ InKIAI プロジェクト @ 宮崎県美郷町 熊本大学副島慶人川村諒 1 実験の目的 従来 信号の受信電波強度 (RSSI:RecevedSgnal StrengthIndcator) により 対象の位置を推定する手法として 無線 LAN の AP(AccessPont) から受信する信号の減衰量をもとに位置を推定する手法が多く検討されている
PowerPoint Presentation
 工学部 6 7 8 9 10 組 ( 奇数学籍番号 ) 担当 : 長谷川英之 情報処理演習 第 7 回 2010 年 11 月 18 日 1 今回のテーマ 1: ポインタ 変数に値を代入 = 記憶プログラムの記憶領域として使用されるものがメモリ ( パソコンの仕様書における 512 MB RAM などの記述はこのメモリの量 ) RAM は多数のコンデンサの集合体 : 電荷がたまっている (1)/ いない
工学部 6 7 8 9 10 組 ( 奇数学籍番号 ) 担当 : 長谷川英之 情報処理演習 第 7 回 2010 年 11 月 18 日 1 今回のテーマ 1: ポインタ 変数に値を代入 = 記憶プログラムの記憶領域として使用されるものがメモリ ( パソコンの仕様書における 512 MB RAM などの記述はこのメモリの量 ) RAM は多数のコンデンサの集合体 : 電荷がたまっている (1)/ いない
カイ二乗フィット検定、パラメータの誤差
 統計的データ解析 008 008.. 林田清 ( 大阪大学大学院理学研究科 ) 問題 C (, ) ( x xˆ) ( y yˆ) σ x πσ σ y y Pabx (, ;,,, ) ˆ y σx σ y = dx exp exp πσx ただし xy ˆ ˆ はyˆ = axˆ+ bであらわされる直線モデル上の点 ( ˆ) ( ˆ ) ( ) x x y ax b y ax b Pabx (,
統計的データ解析 008 008.. 林田清 ( 大阪大学大学院理学研究科 ) 問題 C (, ) ( x xˆ) ( y yˆ) σ x πσ σ y y Pabx (, ;,,, ) ˆ y σx σ y = dx exp exp πσx ただし xy ˆ ˆ はyˆ = axˆ+ bであらわされる直線モデル上の点 ( ˆ) ( ˆ ) ( ) x x y ax b y ax b Pabx (,
intra-mart Accel Platform — IM-共通マスタ スマートフォン拡張プログラミングガイド 初版
 Copyright 2012 NTT DATA INTRAMART CORPORATION 1 Top 目次 1. 改訂情報 2. IM- 共通マスタの拡張について 2.1. 前提となる知識 2.1.1. Plugin Manager 2.2. 表記について 3. 汎用検索画面の拡張 3.1. 動作の概要 3.1.1. 汎用検索画面タブの動作概要 3.2. 実装の詳細 3.2.1. 汎用検索画面タブの実装
Copyright 2012 NTT DATA INTRAMART CORPORATION 1 Top 目次 1. 改訂情報 2. IM- 共通マスタの拡張について 2.1. 前提となる知識 2.1.1. Plugin Manager 2.2. 表記について 3. 汎用検索画面の拡張 3.1. 動作の概要 3.1.1. 汎用検索画面タブの動作概要 3.2. 実装の詳細 3.2.1. 汎用検索画面タブの実装
Information Theory
 前回の復習 講義の概要 chapter 1: 情報を測る... エントロピーの定義 確率変数 X の ( 一次 ) エントロピー M H 1 (X) = p i log 2 p i (bit) i=1 M は実現値の個数,p i は i 番目の実現値が取られる確率 実現値 確率 表 裏 0.5 0.5 H 1 X = 0.5 log 2 0.5 0.5log 2 0.5 = 1bit 1 練習問題の解答
前回の復習 講義の概要 chapter 1: 情報を測る... エントロピーの定義 確率変数 X の ( 一次 ) エントロピー M H 1 (X) = p i log 2 p i (bit) i=1 M は実現値の個数,p i は i 番目の実現値が取られる確率 実現値 確率 表 裏 0.5 0.5 H 1 X = 0.5 log 2 0.5 0.5log 2 0.5 = 1bit 1 練習問題の解答
数値計算法
 数値計算法 008 4/3 林田清 ( 大阪大学大学院理学研究科 ) 実験データの統計処理その 誤差について 母集団と標本 平均値と標準偏差 誤差伝播 最尤法 平均値につく誤差 誤差 (Error): 真の値からのずれ 測定誤差 物差しが曲がっていた 測定する対象が室温が低いため縮んでいた g の単位までしかデジタル表示されない計りで g 以下 計りの目盛りを読み取る角度によって値が異なる 統計誤差
数値計算法 008 4/3 林田清 ( 大阪大学大学院理学研究科 ) 実験データの統計処理その 誤差について 母集団と標本 平均値と標準偏差 誤差伝播 最尤法 平均値につく誤差 誤差 (Error): 真の値からのずれ 測定誤差 物差しが曲がっていた 測定する対象が室温が低いため縮んでいた g の単位までしかデジタル表示されない計りで g 以下 計りの目盛りを読み取る角度によって値が異なる 統計誤差
Microsoft PowerPoint - 10.pptx
 m u. 固有値とその応用 8/7/( 水 ). 固有値とその応用 固有値と固有ベクトル 行列による写像から固有ベクトルへ m m 行列 によって線形写像 f : R R が表せることを見てきた ここでは 次元平面の行列による写像を調べる とし 写像 f : を考える R R まず 単位ベクトルの像 u y y f : R R u u, u この事から 線形写像の性質を用いると 次の格子上の点全ての写像先が求まる
m u. 固有値とその応用 8/7/( 水 ). 固有値とその応用 固有値と固有ベクトル 行列による写像から固有ベクトルへ m m 行列 によって線形写像 f : R R が表せることを見てきた ここでは 次元平面の行列による写像を調べる とし 写像 f : を考える R R まず 単位ベクトルの像 u y y f : R R u u, u この事から 線形写像の性質を用いると 次の格子上の点全ての写像先が求まる
スクールCOBOL2002
 3. 関連資料 - よく使われる機能の操作方法 - (a) ファイルの入出力処理 - 順ファイル等を使ったプログラムの実行 - - 目次 -. はじめに 2. コーディング上の指定 3. 順ファイルの使用方法 4. プリンタへの出力方法 5. 索引ファイルの使用方法 6. 終わりに 2 . はじめに 本説明書では 簡単なプログラム ( ファイル等を使わないプログラム ) の作成からコンパイル 実行までの使用方法は既に理解しているものとして
3. 関連資料 - よく使われる機能の操作方法 - (a) ファイルの入出力処理 - 順ファイル等を使ったプログラムの実行 - - 目次 -. はじめに 2. コーディング上の指定 3. 順ファイルの使用方法 4. プリンタへの出力方法 5. 索引ファイルの使用方法 6. 終わりに 2 . はじめに 本説明書では 簡単なプログラム ( ファイル等を使わないプログラム ) の作成からコンパイル 実行までの使用方法は既に理解しているものとして
アプリケーション インスペクションの特別なアクション(インスペクション ポリシー マップ)
") CHAPTER 2 アプリケーションインスペクションの特別なアクション ( インスペクションポリシーマップ ) モジュラポリシーフレームワークでは 多くのアプリケーションインスペクションで実行される特別なアクションを設定できます サービスポリシーでインスペクションエンジンをイネーブルにする場合は インスペクションポリシーマップで定義されるアクションを必要に応じてイネーブルにすることもできます インスペクションポリシーマップが
CHAPTER 2 アプリケーションインスペクションの特別なアクション ( インスペクションポリシーマップ ) モジュラポリシーフレームワークでは 多くのアプリケーションインスペクションで実行される特別なアクションを設定できます サービスポリシーでインスペクションエンジンをイネーブルにする場合は インスペクションポリシーマップで定義されるアクションを必要に応じてイネーブルにすることもできます インスペクションポリシーマップが
Shareresearchオンラインマニュアル
 Chrome の初期設定 以下の手順で設定してください 1. ポップアップブロックの設定 2. 推奨する文字サイズの設定 3. 規定のブラウザに設定 4. ダウンロードファイルの保存先の設定 5.PDFレイアウトの印刷設定 6. ランキングやハイライトの印刷設定 7. 注意事項 なお 本マニュアルの内容は バージョン 61.0.3163.79 の Chrome を基に説明しています Chrome の設定手順や画面については
Chrome の初期設定 以下の手順で設定してください 1. ポップアップブロックの設定 2. 推奨する文字サイズの設定 3. 規定のブラウザに設定 4. ダウンロードファイルの保存先の設定 5.PDFレイアウトの印刷設定 6. ランキングやハイライトの印刷設定 7. 注意事項 なお 本マニュアルの内容は バージョン 61.0.3163.79 の Chrome を基に説明しています Chrome の設定手順や画面については
図 1 アドインに登録する メニューバーに [BAYONET] が追加されます 登録 : Excel 2007, 2010, 2013 の場合 1 Excel ブックを開きます Excel2007 の場合 左上の Office マークをクリックします 図 2 Office マーク (Excel 20
![図 1 アドインに登録する メニューバーに [BAYONET] が追加されます 登録 : Excel 2007, 2010, 2013 の場合 1 Excel ブックを開きます Excel2007 の場合 左上の Office マークをクリックします 図 2 Office マーク (Excel 20](/thumbs/92/109963601.jpg "図 1 アドインに登録する メニューバーに [BAYONET] が追加されます 登録 : Excel 2007, 2010, 2013 の場合 1 Excel ブックを開きます Excel2007 の場合 左上の Office マークをクリックします 図 2 Office マーク (Excel 20") BayoLink Excel アドイン使用方法 1. はじめに BayoLink Excel アドインは MS Office Excel のアドインツールです BayoLink Excel アドインは Excel から API を利用して BayoLink と通信し モデルのインポートや推論の実行を行います BayoLink 本体ではできない 複数のデータを一度に推論することができます なお現状ではソフトエビデンスを指定して推論を行うことはできません
BayoLink Excel アドイン使用方法 1. はじめに BayoLink Excel アドインは MS Office Excel のアドインツールです BayoLink Excel アドインは Excel から API を利用して BayoLink と通信し モデルのインポートや推論の実行を行います BayoLink 本体ではできない 複数のデータを一度に推論することができます なお現状ではソフトエビデンスを指定して推論を行うことはできません
0 21 カラー反射率 slope aspect 図 2.9: 復元結果例 2.4 画像生成技術としての計算フォトグラフィ 3 次元情報を復元することにより, 画像生成 ( レンダリング ) に応用することが可能である. 近年, コンピュータにより, カメラで直接得られない画像を生成する技術分野が生
 に応用することが可能である. 近年, コンピュータにより, カメラで直接得られない画像を生成する技術分野が生") 0 21 カラー反射率 slope aspect 図 2.9: 復元結果例 2.4 画像生成技術としての計算フォトグラフィ 3 次元情報を復元することにより, 画像生成 ( レンダリング ) に応用することが可能である. 近年, コンピュータにより, カメラで直接得られない画像を生成する技術分野が生まれ, コンピューテーショナルフォトグラフィ ( 計算フォトグラフィ ) と呼ばれている.3 次元画像認識技術の計算フォトグラフィへの応用として,
0 21 カラー反射率 slope aspect 図 2.9: 復元結果例 2.4 画像生成技術としての計算フォトグラフィ 3 次元情報を復元することにより, 画像生成 ( レンダリング ) に応用することが可能である. 近年, コンピュータにより, カメラで直接得られない画像を生成する技術分野が生まれ, コンピューテーショナルフォトグラフィ ( 計算フォトグラフィ ) と呼ばれている.3 次元画像認識技術の計算フォトグラフィへの応用として,
青焼 1章[15-52].indd
![青焼 1章[15-52].indd](/thumbs/86/94313777.jpg "青焼 1章[15-52].indd") 1 第 1 章統計の基礎知識 1 1 なぜ統計解析が必要なのか? 人間は自分自身の経験にもとづいて 感覚的にものごとを判断しがちである 例えばある疾患に対する標準治療薬の有効率が 50% であったとする そこに新薬が登場し ある医師がその新薬を 5 人の患者に使ったところ 4 人が有効と判定されたとしたら 多くの医師はこれまでの標準治療薬よりも新薬のほうが有効性が高そうだと感じることだろう しかし
1 第 1 章統計の基礎知識 1 1 なぜ統計解析が必要なのか? 人間は自分自身の経験にもとづいて 感覚的にものごとを判断しがちである 例えばある疾患に対する標準治療薬の有効率が 50% であったとする そこに新薬が登場し ある医師がその新薬を 5 人の患者に使ったところ 4 人が有効と判定されたとしたら 多くの医師はこれまでの標準治療薬よりも新薬のほうが有効性が高そうだと感じることだろう しかし
JavaプログラミングⅠ
 Java プログラミング Ⅰ 12 回目クラス 今日の講義で学ぶ内容 クラスとは クラスの宣言と利用 クラスの応用 クラス クラスとは 異なる複数の型の変数を内部にもつ型です 直観的に表現すると int 型や double 型は 1 1 つの値を管理できます int 型の変数 配列型は 2 5 8 6 3 7 同じ型の複数の変数を管理できます 配列型の変数 ( 配列変数 ) クラスは double
Java プログラミング Ⅰ 12 回目クラス 今日の講義で学ぶ内容 クラスとは クラスの宣言と利用 クラスの応用 クラス クラスとは 異なる複数の型の変数を内部にもつ型です 直観的に表現すると int 型や double 型は 1 1 つの値を管理できます int 型の変数 配列型は 2 5 8 6 3 7 同じ型の複数の変数を管理できます 配列型の変数 ( 配列変数 ) クラスは double
( 目次 ) 1. XOOPSインストールガイド はじめに 制限事項 サイト初期設定 XOOPSのインストール はじめに データベースの作成 XOOPSのインストール
 1. XOOPSインストールガイド はじめに 制限事項 サイト初期設定 XOOPSのインストール はじめに データベースの作成 XOOPSのインストール") KDDI ホスティングサービス (G120, G200) XOOPS インストールガイド ( ご参考資料 ) rev1.0 KDDI 株式会社 1 ( 目次 ) 1. XOOPSインストールガイド...3 1-1 はじめに...3 1-2 制限事項...3 1-3 サイト初期設定...4 2. XOOPSのインストール...9 3-1 はじめに...9 3-2 データベースの作成...9 3-3 XOOPSのインストール...10
KDDI ホスティングサービス (G120, G200) XOOPS インストールガイド ( ご参考資料 ) rev1.0 KDDI 株式会社 1 ( 目次 ) 1. XOOPSインストールガイド...3 1-1 はじめに...3 1-2 制限事項...3 1-3 サイト初期設定...4 2. XOOPSのインストール...9 3-1 はじめに...9 3-2 データベースの作成...9 3-3 XOOPSのインストール...10
コンピュータグラフィックス基礎 No
 課題 6: モデリング (1) OBJView の動作確認 ( レポートには含めなくてよい ) 次ページ以降の 課題用メモ を参考にして OBJ ファイルを 3D 表示する OBJView を実行し 画面に立体が表示されることを確認するとともに 以下の機能を確認しなさい 左ドラッグによる立体の回転 右ドラッグによる拡大/ 縮小 [v] キーによる頂点の表示 非表示 サンプルに含まれる bunny_3k.obj
課題 6: モデリング (1) OBJView の動作確認 ( レポートには含めなくてよい ) 次ページ以降の 課題用メモ を参考にして OBJ ファイルを 3D 表示する OBJView を実行し 画面に立体が表示されることを確認するとともに 以下の機能を確認しなさい 左ドラッグによる立体の回転 右ドラッグによる拡大/ 縮小 [v] キーによる頂点の表示 非表示 サンプルに含まれる bunny_3k.obj
フローチャート自動生成ツール yflowgen の使い方 目次 1 はじめに 本ツールの機能 yflowgen.exe の使い方 yflowgen.exe の実行方法 制限事項 生成したファイル (gml ファイル形式 ) の開
 の開") フローチャート自動生成ツール yflowgen の使い方 目次 1 はじめに...2 2 本ツールの機能...2 3 yflowgen.exe の使い方...3 3.1 yflowgen.exe の実行方法...3 3.2 制限事項...3 3.3 生成したファイル (gml ファイル形式 ) の開き方...4 3.3.1 yed Graph Editor を使って開く...4 3.3.2 yed
フローチャート自動生成ツール yflowgen の使い方 目次 1 はじめに...2 2 本ツールの機能...2 3 yflowgen.exe の使い方...3 3.1 yflowgen.exe の実行方法...3 3.2 制限事項...3 3.3 生成したファイル (gml ファイル形式 ) の開き方...4 3.3.1 yed Graph Editor を使って開く...4 3.3.2 yed
独立行政法人産業技術総合研究所 PMID-Extractor ユーザ利用マニュアル バイオメディシナル情報研究センター 2009/03/09 第 1.0 版
 独立行政法人産業技術総合研究所 PMID-Extractor ユーザ利用マニュアル バイオメディシナル情報研究センター 2009/03/09 第 1.0 版 目次 1. はじめに... 3 2. インストール方法... 4 3. プログラムの実行... 5 4. プログラムの終了... 5 5. 操作方法... 6 6. 画面の説明... 8 付録 A:Java のインストール方法について... 11
独立行政法人産業技術総合研究所 PMID-Extractor ユーザ利用マニュアル バイオメディシナル情報研究センター 2009/03/09 第 1.0 版 目次 1. はじめに... 3 2. インストール方法... 4 3. プログラムの実行... 5 4. プログラムの終了... 5 5. 操作方法... 6 6. 画面の説明... 8 付録 A:Java のインストール方法について... 11
農業・農村基盤図の大字小字コードXML作成 説明書
 農業 農村基盤図の大字小字コード XML 作成説明書 2007/06/06 有限会社ジオ コーチ システムズ http://www.geocoach.co.jp/ info@geocoach.co.jp 農業 農村基盤図の大字小字コード XML 作成 プログラムについての説明書です バージョン ビルド 1.01 2007/06/06 農業 農村基盤図の大字小字コード XML 作成 は 市区町村 大字
農業 農村基盤図の大字小字コード XML 作成説明書 2007/06/06 有限会社ジオ コーチ システムズ http://www.geocoach.co.jp/ info@geocoach.co.jp 農業 農村基盤図の大字小字コード XML 作成 プログラムについての説明書です バージョン ビルド 1.01 2007/06/06 農業 農村基盤図の大字小字コード XML 作成 は 市区町村 大字
AutoCAD LT2000i
 空間デザイン演習資料 ( 第 10 回 -14 回 ) 課題 国土地理院の基盤地図情報数値標高モデルから東北地方の任意地域の標高データと航空写真を取得し, 以下の設計条件を満足する道路設計を行いなさい. また, 走行シミュレーションのアニメーションを作成しなさい.(Civil3D の新規 国土交通省仕様 100m 測点.dwt を用いて設計をすること ) 注意 : これまでの配布資料を忘れずに持参しなさい.
空間デザイン演習資料 ( 第 10 回 -14 回 ) 課題 国土地理院の基盤地図情報数値標高モデルから東北地方の任意地域の標高データと航空写真を取得し, 以下の設計条件を満足する道路設計を行いなさい. また, 走行シミュレーションのアニメーションを作成しなさい.(Civil3D の新規 国土交通省仕様 100m 測点.dwt を用いて設計をすること ) 注意 : これまでの配布資料を忘れずに持参しなさい.
様々なミクロ計量モデル†
 担当 : 長倉大輔 ( ながくらだいすけ ) この資料は私の講義において使用するために作成した資料です WEB ページ上で公開しており 自由に参照して頂いて構いません ただし 内容について 一応検証してありますが もし間違いがあった場合でもそれによって生じるいかなる損害 不利益について責任を負いかねますのでご了承ください 間違いは発見次第 継続的に直していますが まだ存在する可能性があります 1 カウントデータモデル
担当 : 長倉大輔 ( ながくらだいすけ ) この資料は私の講義において使用するために作成した資料です WEB ページ上で公開しており 自由に参照して頂いて構いません ただし 内容について 一応検証してありますが もし間違いがあった場合でもそれによって生じるいかなる損害 不利益について責任を負いかねますのでご了承ください 間違いは発見次第 継続的に直していますが まだ存在する可能性があります 1 カウントデータモデル
生命情報学
 生命情報学 5 隠れマルコフモデル 阿久津達也 京都大学化学研究所 バイオインフォマティクスセンター 内容 配列モチーフ 最尤推定 ベイズ推定 M 推定 隠れマルコフモデル HMM Verアルゴリズム EMアルゴリズム Baum-Welchアルゴリズム 前向きアルゴリズム 後向きアルゴリズム プロファイル HMM 配列モチーフ モチーフ発見 配列モチーフ : 同じ機能を持つ遺伝子配列などに見られる共通の文字列パターン
生命情報学 5 隠れマルコフモデル 阿久津達也 京都大学化学研究所 バイオインフォマティクスセンター 内容 配列モチーフ 最尤推定 ベイズ推定 M 推定 隠れマルコフモデル HMM Verアルゴリズム EMアルゴリズム Baum-Welchアルゴリズム 前向きアルゴリズム 後向きアルゴリズム プロファイル HMM 配列モチーフ モチーフ発見 配列モチーフ : 同じ機能を持つ遺伝子配列などに見られる共通の文字列パターン
EBNと疫学
 推定と検定 57 ( 復習 ) 記述統計と推測統計 統計解析は大きく 2 つに分けられる 記述統計 推測統計 記述統計 観察集団の特性を示すもの 代表値 ( 平均値や中央値 ) や ばらつきの指標 ( 標準偏差など ) 図表を効果的に使う 推測統計 観察集団のデータから母集団の特性を 推定 する 平均 / 分散 / 係数値などの推定 ( 点推定 ) 点推定値のばらつきを調べる ( 区間推定 ) 検定統計量を用いた検定
推定と検定 57 ( 復習 ) 記述統計と推測統計 統計解析は大きく 2 つに分けられる 記述統計 推測統計 記述統計 観察集団の特性を示すもの 代表値 ( 平均値や中央値 ) や ばらつきの指標 ( 標準偏差など ) 図表を効果的に使う 推測統計 観察集団のデータから母集団の特性を 推定 する 平均 / 分散 / 係数値などの推定 ( 点推定 ) 点推定値のばらつきを調べる ( 区間推定 ) 検定統計量を用いた検定
Medical3
 1.4.1 クロス集計表の作成 -l m 分割表 - 3つ以上のカテゴリを含む変数を用いて l mのクロス集計表による分析を行います この例では race( 人種 ) によってlow( 低体重出生 ) に差が認められるかどうかを分析します 人種には3つのカテゴリ 低体重出生には2つのカテゴリが含まれています 2つの変数はともにカテゴリ変数であるため クロス集計表によって分析します 1. 分析メニュー
1.4.1 クロス集計表の作成 -l m 分割表 - 3つ以上のカテゴリを含む変数を用いて l mのクロス集計表による分析を行います この例では race( 人種 ) によってlow( 低体重出生 ) に差が認められるかどうかを分析します 人種には3つのカテゴリ 低体重出生には2つのカテゴリが含まれています 2つの変数はともにカテゴリ変数であるため クロス集計表によって分析します 1. 分析メニュー
PowerPoint プレゼンテーション
 V1 次世代シークエンサ実習 II 本講義にあたって 代表的な解析の流れを紹介します 論文でよく使用されているツールを使用します コマンドを沢山実行します スペルミスが心配な方は コマンド例がありますのでコピーして実行してください /home/admin1409/amelieff/ngs/reseq_command.txt マークのコマンドは実行してください 実行が遅れてもあせらずに 応用や課題の間に追い付いてください
V1 次世代シークエンサ実習 II 本講義にあたって 代表的な解析の流れを紹介します 論文でよく使用されているツールを使用します コマンドを沢山実行します スペルミスが心配な方は コマンド例がありますのでコピーして実行してください /home/admin1409/amelieff/ngs/reseq_command.txt マークのコマンドは実行してください 実行が遅れてもあせらずに 応用や課題の間に追い付いてください
KEGG.ppt
 1 2 3 4 KEGG: Kyoto Encyclopedia of Genes and Genomes http://www.genome.jp/kegg/kegg2.html http://www.genome.jp/kegg/kegg_ja.html 5 KEGG PATHWAY 生体内(外)の分子間ネットワーク図 代謝系 12カテゴリ 中間代謝 二次代謝 薬の 代謝 全体像 制御系 20カテゴリ
1 2 3 4 KEGG: Kyoto Encyclopedia of Genes and Genomes http://www.genome.jp/kegg/kegg2.html http://www.genome.jp/kegg/kegg_ja.html 5 KEGG PATHWAY 生体内(外)の分子間ネットワーク図 代謝系 12カテゴリ 中間代謝 二次代謝 薬の 代謝 全体像 制御系 20カテゴリ