機能ゲノム学(第6回)
|
|
|
- まとも のあき
- 4 years ago
- Views:
Transcription
1 R でトランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp 1
 2002/4/1~ 産総研")

2 自己紹介 2002 年 3 月 東京大学 大学院農学生命科学研究科博士課程修了 学位論文 : cdna マイクロアレイを用いた遺伝子発現解析手法の開発 ( 指導教官 : 清水謙多郎教授 ) 2002/4/1~ 産総研 生命情報科学研究センター (CBRC) 産総研特別研究員 2003/11/1~ 放医研 先端遺伝子発現研究センター研究員 2005/2/16~ 東京大学 大学院農学生命科学研究科特任助手 2007/4/1~ 現在 東京大学 大学院農学生命科学研究科特任助教 アグリバイオインフォマティクスプログラム 2

3 様々な Motivation ~ の原因遺伝子 ( ガン関連遺伝子とか ) を同定したい FASTQ 以降の一通りの解析ができるようになりたい (Windows の )R でできることとできないこと モデル生物と非モデル生物の解析戦略の違い 倍率変化で解析 vs. 分布を使って解析 いろんな R パッケージがあるけれど RNA-seq で二つのサンプルを比較し 発現変動遺伝子同定までを行うまでの流れを一通り紹介 A 群腎臓正常組織 wildtype B 群肝臓腫瘍組織 mutant 3

4 データ解析のスタート地点 NGS から得られた FASTQ 形式ファイル データ取得完了! A1.fq A2.fq B1.fq B2.fq なんじゃこの変な記号は! 何をどうすれば... 4
5 比較トランスクリプトーム解析の流れ 複数の FASTQ ファイル Linux マシン リファレンス配列の作成 A1.fq, A2.fq, B1.fq, B2.fq 複数サンプルの混合アセンブルにより RefSeq のような転写物配列集合 (multi-fasta ファイル ) を得るイメージ マッピング どの転写物にどれだけの数のリードがマップされたかという いわゆる 遺伝子発現行列 を得るイメージ データ解析 発現変動遺伝子のリストアップや 作図など すべてが (Windows の )R でできるわけではありません 5

6 Linux マシン使用部分の解決策 自前で大容量メモリ計算サーバ (Linux) を購入し 必要なソフトのインストールからスタート 特徴 : 難易度は高いが思い通りの解析が可能 Linux サーバをもつバイオインフォ系の人にお願いする 特徴 : 気軽に頼める知り合いがいればいいが その人次第 DDBJ Read Annotation Pipeline を利用 特徴 : 一番お手軽な選択肢だが サポートしているプログラムのみ データ登録が前提?! だが 手取り足取り丁寧に教えてくれるので個人的にはこちらを推奨 自分の負担も減るし 6

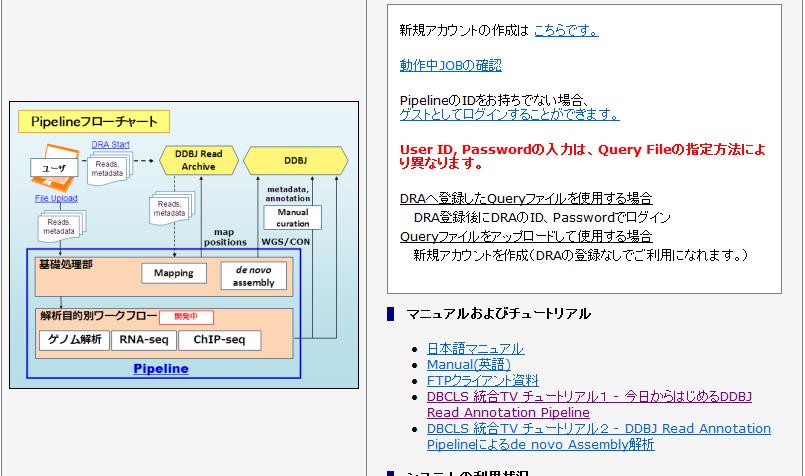
7 7
8 比較トランスクリプトーム解析の流れ 複数の FASTQ ファイル Linux マシン リファレンス配列の作成 クオリティチェック アセンブル結果 (multi-fasta) ファイルから平均長やトータルの長さなどの基本情報を抽出 マッピング マッピング結果 (BED 形式 ) ファイルを入力として 転写物ごとのマップされたリード数をカウント データ解析 発現変動遺伝子のリストアップや 作図など 大規模計算部分以外は一通りできます 8

9 参考ウェブページ 9
10 比較トランスクリプトーム解析の流れ 複数の FASTQ ファイル クオリティチェック リファレンス配列の作成 アセンブル結果 (multi-fasta) ファイルから平均長やトータルの長さなどの基本情報を抽出 マッピング マッピング結果 (BED 形式 ) ファイルを入力として 転写物ごとのマップされたリード数をカウント データ解析 発現変動遺伝子のリストアップや 作図など 10

11 データのクオリティチェック デスクトップにある フォルダ中に SRR fastq というファイルが存在する という前提 11

12 R の起動 デスクトップにある フォルダ中のファイルを解析 12
")

13 作業ディレクトリ (= フォルダ ) の変更
14 getwd() と打ち込んで確認 14
 1 2 1")

15 コピー & ペースト ( こぴぺ ) 一連のコマンド群をコピーして 2R Console 画面上でペースト 15


16 html レポートが作成される R ではうまくいくが R ではエラーがでます 16
17 html レポートが作成される (R では ) ポジションごとのクオリティスコアなどの情報が得られます 17

)%%%++)(%%%%).")
18 FASTQ 形式 ( と FASTA 形式 ) FASTA 形式 > ではじまる一行の description 行 と 配列情報 からなる形式 NGS の read 長は短いので 実質的に一つのリードを二行で表現 >SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT FASTQ 形式 一行目 ではじまる一行の description 行 二行目 : 配列情報 三行目 : + からはじまる一行 ( の description 行 ) 四行目 : GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT +!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC

19 塩基配列のクオリティ情報といえば Phred スコア Phred というベースコールプログラムから得られる Quality Value(QV 値 ) のこと なぜ FASTQ 形式では Phred スコアそのものでクオリティ情報を表現しないの? 19

")
20 理由 :( 容量 ) 節約のため Cock et al., Nucleic Acids Res., 38: , 2010 FASTQ 形式中のクオリティ情報部分 Phred スコア (QUAL 形式 ) Phred スコアが X の場合 ASCII (X+33) に対応する文字コードを割り当てる 20
21 R の現実 (R で ) 塩基配列解析 のウェブページは常に最新の情報が記載されているわけではない 昔のバージョンだとうまくいくが 最新版だとエラーがでる こともある R のバージョンを上げてから昔作ったスクリプトを実行しようとすると そんな関数ない と文句を言われた よく調べてみると関数名が変わっていた 例 :DESeq パッケージ中の estimatevariancefunctions estimatedispersions DEGseq のスクリプトがうまく動かなくなっている R ( ) R ( ) での体験談 21
 で現在利用している R のバージョンを確認 手元にある印刷物のマニュアルは古いものでは?! 22")
22 R の現実 と対処法 ぐぐる ( qrqc error などで検索) Rのバージョンを最新版 ( または古いもの ) に変更 sessioninfo() で現在利用している R のバージョンを確認 手元にある印刷物のマニュアルは古いものでは?! 22

23 R の昔のバージョンのインストール (R で ) マイクロアレイデータ解析 のウェブページのほうです 23
24 比較トランスクリプトーム解析の流れ 複数の FASTQ ファイル クオリティチェック リファレンス配列の作成 アセンブル結果 (multi-fasta) ファイルから平均長やトータルの長さなどの基本情報を抽出 マッピング マッピング結果 (BED 形式 ) ファイルを入力として 転写物ごとのマップされたリード数をカウント データ解析 発現変動遺伝子のリストアップや 作図など 24
25 リファレンス配列について Q: マッピングに使うリファレンス配列は? A: 好きなものを使ってください ゲノム配列でもトランスクリプトーム配列でも結構です Q: どこから取得できるんですか? A: UCSC Sequence and Annotation Downloads などから取得できます ( アノテーション情報も ) 以下はほんの一例 ヒト全ゲノム配列の場合 ftp://genome-ftp.cse.ucsc.edu/goldenpath/hg19/bigzips/hg19.2bit ヒトトランスクリプトーム配列 (RefSeq mrna) の場合 ftp://genome-ftp.cse.ucsc.edu/goldenpath/hg19/bigzips/refmrna.fa.gz ヒトアノテーション情報の場合

 symbol 染色体名転写開始位置 name CDS")
26 こんな感じ ヒトトランスクリプトーム配列 ヒトゲノム配列 1-22 番染色体 +X+Y 約 6200 万行のファイル 約 3GB のサイズ 46,XXX mrna sequences ftp://ftp.ncbi.nih.gov/refseq/h_sapiens/ mrna_prot/human.rna.fna.gz アノテーション情報ファイル (refflat 形式 ) symbol 染色体名転写開始位置 name CDS start Exon 数 strand 転写終結位置 CDS end 26
27 非モデル生物の場合 手持ちの RNA-Seq データのみ 2010 年頃から提供されはじめた de novo transcriptome assembly 用のプログラム (Trinity や Trans-ABySS など ; もちろん Linux 用です ) を利用すればトランスクリプトームの配列セット (RefSeq のようなイメージ ) を得ることができます 入力 :RNA-Seq データ出力 : コンティグ ( 転写物配列 ) >read1 GGGGTTCAAAGCAGTATCGATCAAATAGTA >read2 GTTCAAAGCAGTATCGATCAAATAGTAAAT >read3 ACGATGCAGCCTTAACGATGGTCCACAATT >read4 >contig1(transcript1) GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAA CTCACAGTTTGGAGCTTATCAGTCAA >contig2(transcript2) ACGATGCAGCCTTAACGATGGTCCACAATTATCGGGAATCA >contig3(transcript3) 27
28 非モデル生物の場合 手持ちの RNA-Seq データのみでアセンブルを行う場合は paired-end のデータが基本 ペアードエンド法断片配列の両末端が数百塩基以内の対の二種類の配列が得られる A1: A1_1.fq A1_2.fq A2: A2_1.fq A2_2.fq シングルエンド法 約 塩基 B1: B1_1.fq B1_2.fq B2: B2_1.fq B2_2.fq read_1.fq read_2.fq 二つのファイルを入力として比較対象サンプルを混合したデータのアセンブル リファレンストランスクリプトーム配列の取得 28
29 非モデル生物の場合 Trinity (Grabherr et al., Nat. Biotechnol., 2011) 実行プログラムの一例 nohup Trinity.pl --seqtype fq --left read_1.fq --right read_2.fq -- run_butterfly --bflyheapspace 180G --CPU 2 --bfly_opts "-V stderr" --run_allpathslg_error_correction --output hoge & アセンブルが終了すると hoge というディレクトリ中に Trinity.fasta という multi-fasta ファイルが得られる 29
30 比較トランスクリプトーム解析の流れ 複数の FASTQ ファイル クオリティチェック リファレンス配列の作成 アセンブル結果 (multi-fasta) ファイルから平均長やトータルの長さなどの基本情報を抽出 マッピング マッピング結果 (BED 形式 ) ファイルを入力として 転写物ごとのマップされたリード数をカウント データ解析 発現変動遺伝子のリストアップや 作図など 30

31 multi-fasta 形式ファイルからの情報抽出 一連のコマンド群をコピーして 2R Console 画面上でペースト 31


32 multi-fasta 形式ファイルからの情報抽出 1 出力ファイル名として指定したもの (hoge.txt) が フォルダ中に作成される ( はず ) 32


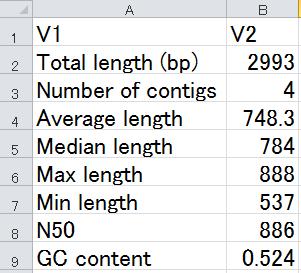
33 練習 中にある practice1.txt 中の記述を変更して Trinity.fasta ファイルに対して同様の解析を行い 結果を hoge1.txt に出力せよ 33
34 multi-fasta 形式ファイルからの情報抽出 2 配列ごとの GC 含量を計算したいとき 34


35 練習 中にある practice2.txt 中の記述を変更して Trinity.fasta ファイルに対して同様の解析を行い 結果を hoge2.txt に出力せよ 35

36 multi-fasta 形式ファイルからの情報抽出 3 Trinity.fasta に対して 600bp 以上のもののみ抽出してみよう 36

37 multi-fasta 形式ファイルからの情報抽出 4 FPKM 値 : 配列長補正ずみの発現量に相当する値 Trinity の最近のバージョンでは FPKM 値を出力しなくなったようですね 37


38 multi-fasta 形式ファイルからの情報抽出 4 Trinity.fasta FPKM 値をもとにサンプル内の転写物間の発現レベルの大小を議論可能 サンプル間の比較には使えない ( といわれている ) 38
39 比較トランスクリプトーム解析の流れ 複数の FASTQ ファイル クオリティチェック リファレンス配列の作成 アセンブル結果 (multi-fasta) ファイルから平均長やトータルの長さなどの基本情報を抽出 マッピング マッピング結果 (BED 形式 ) ファイルを入力として 転写物ごとのマップされたリード数をカウント データ解析 発現変動遺伝子のリストアップや 作図など 39

40 マッピングの基本的なイメージ 基本的なマッピングプログラム (bowtie など ) を用いた場合 リファレンス配列 : ゲノム count T1 サンプルの RNA-Seq データ mapping 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 リファレンス配列 : トランスクリプトーム count 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 40
41 解析の目的に応じてプログラムを使い分け ゲノム配列既知で遺伝子構造を知りたいような場合 Cufflinks (Trapnell et al., Nat. Biotechnol., 2010) ARTADE2 (Kawaguchi et al., Bioinformatics, 2012) ( ゲノム配列の有無に関わらず )RefSeq のようなトランスクリプトーム配列にマッピングして比較トランスクリプトーム解析を行いたい場合 Bowtie (Langmead et al., Genome Biol., 2009) BWA (Li and Durbin, Bioinformatics, 2009) Cufflinks 周辺は専門外です 41
42 マッピング結果の出力ファイル形式 ( ゲノム配列の場合 ) どの染色体上のどの位置に ( どのリードが ) マッピングされたか あるいは ( トランスクリプトーム配列の場合 ) どの転写物配列上のどの位置に ( どのリードが ) マッピングされたかを表すファイル形式 ( フォーマット ) は複数あります : BED (Browser Extensible Data) format BEDtools (Quinlan et al., Bioinformatics, 26: , 2010) GFF (General Feature Format) format SAM (Sequence Alignment/Map) format SAMtools (Li et al., Bioinformatics, 25: , 2009) マッピング結果ファイルから どうやって転写物ごとのマップされたリード数をカウントするのか? 42


43 BED 形式 43

44 BED 形式 あるトランスクリプトーム配列 (RefSeq) にマップした結果 転写物 ID Start End 転写物 ID ごとの出現数 = マップされたリード数 44
45 比較トランスクリプトーム解析の流れ 複数の FASTQ ファイル クオリティチェック リファレンス配列の作成 アセンブル結果 (multi-fasta) ファイルから平均長やトータルの長さなどの基本情報を抽出 マッピング マッピング結果 (BED 形式 ) ファイルを入力として 転写物ごとのマップされたリード数をカウント データ解析 発現変動遺伝子のリストアップや 作図など 45


46 R を用いてコピペでマップされたリード数情報を得ることができます output3.txt 46
47 データ解析の前に 研究目的 ( と手持ちのデータ ) をおさらい 一つのサンプル内でどの転写物 (or 遺伝子 ) の発現レベルが高いか低いかを調べたい場合 RPKM や FPKM などの 転写物の長さを考慮して正規化されたデータ で解析 トータルのリード数を補正する必要はないがやってもよい 遺伝子間の発現レベルの大小関係を調べたいだけなので 解析データを定数倍したところで何ら影響を与えないから サンプル間比較 (sample A vs. B など ) で 発現変動遺伝子 ( Differentially Expressed Genes; DEGs) を調べたい場合 トータルのリード数を補正したデータ で解析 正確には サンプル間で発現変動していない遺伝子 (non-degs) ができるだけ発現変動していないと判定されるように正規化したデータ 既存の R パッケージを用いて解析を行う場合には ( 整数値のみからなる ) 生のリードカウントデータ を入力とし 内部的に上記正規化を行う 研究目的によってやっていい正規化とやってはいけない ( と言われている ) 正規化がある 47
48 比較トランスクリプトーム解析の流れ 複数の FASTQ ファイル クオリティチェック リファレンス配列の作成 アセンブル結果 (multi-fasta) ファイルから平均長やトータルの長さなどの基本情報を抽出 マッピング マッピング結果 (BED 形式 ) ファイルを入力として 転写物ごとのマップされたリード数をカウント データ解析 発現変動遺伝子のリストアップや 作図など 48
49 二群間比較用 R パッケージ DEGseq (Wang et al., Bioinformatics, 26: , 2010) ポワソン分布 (variance = mean) を仮定しているためばらつきを過少評価 edger (Robinson et al., Bioinformatics, 26: , 2010) 正規化法 :TMM 法 負の二項分布 (variance > mean) を仮定 mean のみのパラメータを用いて現実のばらつきを表現 DESeq (Anders and Huber, Genome Biol., 11: R106, 2010) 正規化法 :RLE 法 (relative log expression) edger のモデルをさらに拡張 ( しているらしい ) bayseq (Hardcastle and Kelly, BMC Bioinformatics, 11:422, 2010) 正規化法 :RPM ( たぶん ) 配列の長さ情報を与えるオプションがある データセット中に占める DEG の割合 (P DEG ) を一意に返す NBPSeq (Di et al., SAGMB, 10:24, 2011) 入力 : 生のリードカウントからなる遺伝子発現行列出力 : 遺伝子ごとの発現変動の度合い (p 値など ) 49
50 理想的な実験デザイン ( 二群間比較 ) サンプル A vs. B の比較 (Kidney vs. Liver; 腎臓 vs. 肝臓 ) 生のリードカウントのデータ ( 整数値 ) Biological replicates のデータ生物学的なばらつき ( 個体間の違い ) を考慮すべし A1: ある生物の腎臓 A2: 同じ生物種の別個体の腎臓 A3: 同じ生物種のさらに別個体の腎臓 B1: ある生物の肝臓 B2: 同じ生物種の別個体の肝臓 50
51 分布の話 例題 :Marioni et al., Genome Res., 18: , 2008 のデータ ( の一部 ) kidney( 腎臓 ) liver( 肝臓 ) Technical replicates のデータサンプル内の技術的なばらつき ( 例 : レーン間の違い ) の度合いを調べるためのデータであり このようなデータで二群間比較し 発現変動遺伝子がどの程度あるかといった数に関する議論は無意味解析例 : アリエナイ?! 数 (50% とか ) が発現変動遺伝子として検出される 理由 :Biological variation > Technical variation 51

 kidney( 腎臓 ) RPM 正規化")
52 分布の話 例題 :Marioni et al., Genome Res., 18: , 2008 のデータ ( の一部 ) kidney( 腎臓 ) RPM 正規化 1,000,000 12,685 1,804,
VARIANCE はその MEAN で説明可能である VARIANCE MEAN ポアソン分布に従う")
53 分布の話 例題 :Marioni et al., Genome Res., 18: , 2008 のデータ ( の一部 ) kidney( 腎臓 ) adjusted R-squared: y = x y = a + bx Technical replicates のデータは : ( 遺伝子の )VARIANCE はその MEAN で説明可能である VARIANCE MEAN ポアソン分布に従う ポアソンモデルが適用可能 53
54 分布の話 生物アイコン ( 例題 :Cumbie et al., PLoS ONE, 6: e25279, 2011 のデータ ( の一部 ) Arabidopsis( シロイヌナズナ ) adjusted R-squared: y = a + bx y = x Biological replicates のデータは : VARIANCE > MEAN 負の二項 (NB) 分布に従う NB モデルが適用可能 54
55 なぜ沢山の方法が存在しうるのか? DEGseq (Wang et al., Bioinformatics, 26: , 2010) ポワソン分布 (variance = mean) を仮定しているためばらつきを過少評価 edger (Robinson et al., Bioinformatics, 26: , 2010) 正規化法 :TMM 法 負の二項分布 (variance > mean) を仮定 DESeq (Anders and Huber, Genome Biol., 11: R106, 2010) 正規化法 :RLE 法 (relative log expression) edgerのモデルをさらに拡張 ( しているらしい ) bayseq (Hardcastle and Kelly, BMC Bioinformatics, 11:422, 2010) 正規化法 :RPM ( たぶん ) 配列の長さ情報を与えるオプションがある データセット中に占めるDEGの割合 (P DEG ) を一意に返す NBPSeq (Di et al., SAGMB, 10:24, 2011) VAR (1 ) VAR (1 ) Ans. Variance と Mean の関係を表現する手段が沢山あるから VAR (1 1 ) VAR 55
56 edger を使ってみる 例題 :Marioni et al., Genome Res., 18: , 2008 のデータ kidney( 腎臓 ) liver( 肝臓 ) ファイル名 :SupplementaryTable2_changed.txt 内容 :A 群が最初の 5 列 B 群が残りの 5 列のデータ解析結果を hoge2.txt という名前でファイルに出力したい 56

57 edger を使ってみる ファイル名 :SupplementaryTable2_changed.txt 内容 :A 群が最初の 5 列 B 群が残りの 5 列のデータ解析結果を hoge2.txt という名前でファイルに出力したい 57


58 edger を使ってみる R 上でスクリプトをコピペ! ( エラーメッセージが出ていなければ hoge2.txt というファイルができているはず ) 58

59 edger を使ってみる 一番右側の数値が False Discovery Rate (FDR) この列 (O 列 ) で昇順にソートすれば任意の閾値を満たす遺伝子数がわかる 19,785 個が FDR < 0.01 を満たす 21,291 個が FDR < 0.05 を満たす 59
")
60 edger を使ってみる Top-ranked gene の生リードカウントを眺めても確かに発現変動 (Kidney << Liver) していることが分かる 60


61 edger を使ってみる M-A plot を描画 (FDR < 0.01 を満たすものを赤色で表示 ) hoge2.png 19,794 個 ( 全遺伝子数のうち約 62% が FDR < 0.01 を満たす ) 61
 hoge2.")
 このやり方はダメなんです")
62 edger を使ってみる M-A plot を描画 (2 倍以上発現変動しているものを赤色で表示 ) hoge2.png 個 ( 全遺伝子数のうち約 37% が 2 倍以上発現変動している ) このやり方はダメなんです 62
63 倍率変化がだめな理由をデモ 例題 :Marioni et al., Genome Res., 18: , 2008 のデータ kidney( 腎臓 ) liver( 肝臓 ) 発現変動遺伝子がないデータで二群間比較をしてみる A 群 B 群 63
64 倍率変化がだめな理由をデモ 例題 :Marioni et al., Genome Res., 18: , 2008 のデータ ( の一部 ) (A1, A2) vs. (A3, A4) の二群間比較結果 edger で FDR < 0.01 を満たすものは 0 個 (edger で )2 倍以上発現変動しているものは 3814 個 Rcode_edgeR_tech_rep_fdr001.txt Rcode_edgeR_tech_rep_fc2.txt 低発現領域で log 比が大きくなる現象をうまくモデル化することが重要 64

65 Top 400 Top 2000 低い 全体的な発現レベル 高い こんな感じでランキングすることが重要です 65
66 26,221 genes Biological replicates の 3 vs. 3 サンプル 例題 :Cumbie et al., PLoS ONE, 6: e25279, 2011 の Arabidopsis データ A 群 B 群 data_arab.txt オリジナルは AT4G32850 のものが重複して存在していたため 行目のデータを予め除去している 66
 で実行")
67 edger を default の手順 (edger/default) で実行 67


68 edger を default の手順 (edger/default) で実行 B 群で高発現 A 群で高発現 68


69 サンプル間クラスタリングも重要です 69

70 サンプル間クラスタリングも重要です データ中に発現変動遺伝子がありそうかどうかはクラスタリング結果を眺めるだけでかなりわかる 70
")
71 東大生以外の方も受講可能です ( 来年度もやります ) 71
(H21-23 年度 ): マイクロアレイ解析の再現性 感度 特異度を飛躍的に向上させるデータ解析手法の開発 ( 代表 ) 新学術領域研究 ( 研究領域提案型 )(H22 年度 -): 非モデル生物におけるゲノム解析法の確立 ( 分担 ; 研究代表者 : 西山智明 )")
72 謝辞 共同研究者清水謙多郎先生 ( 東京大学 ) 嶋田透先生 ( 東京大学 ) 西山智明先生 ( 金沢大学 ) 勝間進先生 ( 東京大学 ) 河岡慎平博士 ( 東京大学 ) 末次克行先生 ( 農業生物資源研究所 ) 上樂明也先生 ( 農業生物資源研究所 ) グラント 若手研究 (B)(H21-23 年度 ): マイクロアレイ解析の再現性 感度 特異度を飛躍的に向上させるデータ解析手法の開発 ( 代表 ) 新学術領域研究 ( 研究領域提案型 )(H22 年度 -): 非モデル生物におけるゲノム解析法の確立 ( 分担 ; 研究代表者 : 西山智明 ) 72
機能ゲノム学(第6回)
") RNA-Seq データ解析リテラシー 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 2009 年ごろの私 次世代シーケンサー (NGS) 解析についての認識 単に短い塩基配列が沢山あるだけでしょ 得られる配列データって
RNA-Seq データ解析リテラシー 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 2009 年ごろの私 次世代シーケンサー (NGS) 解析についての認識 単に短い塩基配列が沢山あるだけでしょ 得られる配列データって
機能ゲノム学(第6回)
") RNA-Seqデータ解析における正規化法の選択 :RPKM 値でサンプル間比較は危険?! 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 よりよい正規化法とは? その正規化法によって得られたデータを用いて発現変動の度合いでランキングしたときに
RNA-Seqデータ解析における正規化法の選択 :RPKM 値でサンプル間比較は危険?! 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 よりよい正規化法とは? その正規化法によって得られたデータを用いて発現変動の度合いでランキングしたときに
ゲノム情報解析基礎 ~ Rで塩基配列解析 ~
 ネット接続できないヒトも ダブルクリックでローカルに r_seq.html を起動可能です 実習は デスクトップ上にある hoge フォルダの中身が以下の状態を想定して行います (R で ) 塩基配列解析の利用法 : GC 含量計算から発現変動解析まで東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
ネット接続できないヒトも ダブルクリックでローカルに r_seq.html を起動可能です 実習は デスクトップ上にある hoge フォルダの中身が以下の状態を想定して行います (R で ) 塩基配列解析の利用法 : GC 含量計算から発現変動解析まで東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
機能ゲノム学(第6回)
") トランスクリプトーム解析の今昔 なぜマイクロアレイ? なぜRNA-Seq? 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 Contents トランスクリプトーム解析の概要 各手法の長所 短所 マイクロアレイ
トランスクリプトーム解析の今昔 なぜマイクロアレイ? なぜRNA-Seq? 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 Contents トランスクリプトーム解析の概要 各手法の長所 短所 マイクロアレイ
基本的な利用法
 (R で ) 塩基配列解析 基本的な利用法 Macintosh 版 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける 1. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール 2. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握
(R で ) 塩基配列解析 基本的な利用法 Macintosh 版 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける 1. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール 2. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握
Rでゲノム・トランスクリプトーム解析
 R でゲノム トランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 自己紹介 1995 年 3 月 高知工業高等専門学校 工業化学科卒業 1997 年 3 月 東京農工大学 工学部 物質生物工学科卒業
R でゲノム トランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 自己紹介 1995 年 3 月 高知工業高等専門学校 工業化学科卒業 1997 年 3 月 東京農工大学 工学部 物質生物工学科卒業
NGSデータ解析入門Webセミナー
 NGS データ解析入門 Web セミナー : RNA-Seq 解析編 1 RNA-Seq データ解析の手順 遺伝子発現量測定 シークエンス マッピング サンプル間比較 機能解析など 2 CLC Genomics Workbench 使用ツール シークエンスデータ メタデータのインポート NGS data import Import Metadata クオリティチェック Create Sequencing
NGS データ解析入門 Web セミナー : RNA-Seq 解析編 1 RNA-Seq データ解析の手順 遺伝子発現量測定 シークエンス マッピング サンプル間比較 機能解析など 2 CLC Genomics Workbench 使用ツール シークエンスデータ メタデータのインポート NGS data import Import Metadata クオリティチェック Create Sequencing
Rでトランスクリプトーム解析
 R でトランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 自己紹介 1995 年 3 月 高知工業高等専門学校 工業化学科卒業 1997 年 3 月 東京農工大学 工学部 物質生物工学科卒業
R でトランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 自己紹介 1995 年 3 月 高知工業高等専門学校 工業化学科卒業 1997 年 3 月 東京農工大学 工学部 物質生物工学科卒業
特論I
 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 4 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp 1 前回の課題と正答 アダプター配列除去前後の small RNA-seq データをカイコゲノムにマップし マップ率 ( マップされたリード数
講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 4 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp 1 前回の課題と正答 アダプター配列除去前後の small RNA-seq データをカイコゲノムにマップし マップ率 ( マップされたリード数
シーケンサー利用技術講習会 第10回 サンプルQC、RNAseqライブラリー作製/データ解析実習講習会
 シーケンサー利用技術講習会 第 10 回サンプル QC RNAseq ライブ ラリー作製 / データ解析実習講習会 理化学研究所ライフサイエンス技術基盤研究センターゲノムネットワーク解析支援施設田上道平 次世代シーケンサー Sequencer File Format Output(Max) Read length Illumina Hiseq2500 Fastq 600 Gb 100 bp Life
シーケンサー利用技術講習会 第 10 回サンプル QC RNAseq ライブ ラリー作製 / データ解析実習講習会 理化学研究所ライフサイエンス技術基盤研究センターゲノムネットワーク解析支援施設田上道平 次世代シーケンサー Sequencer File Format Output(Max) Read length Illumina Hiseq2500 Fastq 600 Gb 100 bp Life
Rインストール手順
 R 本体は最新のリリース版 R パッケージは 必要最小限プラスアルファ の推奨インストール手順を示します R 本体とパッケージのインストール Windows 版 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/
R 本体は最新のリリース版 R パッケージは 必要最小限プラスアルファ の推奨インストール手順を示します R 本体とパッケージのインストール Windows 版 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/
PowerPoint プレゼンテーション
 バイオインフォマティクス 講習会 V 事前準備 が完了されている方は コンテナの起動 ファイルのコピー (Windows) まで 進めておいてください メニュー 1. 環境構築の確認 2. 基本的なLinuxコマンド 3. ツールのインストール 4. NGSデータの基礎知識と前処理 5. トランスクリプトのアッセンブル 6. RNA-seqのリファレンスcDNAマッピングとFPKM 算出 7. RNA-seqのリファレンスゲノムマッピングとFPKM
バイオインフォマティクス 講習会 V 事前準備 が完了されている方は コンテナの起動 ファイルのコピー (Windows) まで 進めておいてください メニュー 1. 環境構築の確認 2. 基本的なLinuxコマンド 3. ツールのインストール 4. NGSデータの基礎知識と前処理 5. トランスクリプトのアッセンブル 6. RNA-seqのリファレンスcDNAマッピングとFPKM 算出 7. RNA-seqのリファレンスゲノムマッピングとFPKM
NGS速習コース
 バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-3. R 各種パッケージ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-3. R 各種パッケージ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
機能ゲノム学(第6回)
") バイオインフォマティクス次世代シーケンサー (NGS) 編 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 バイオインフォマティクス人材育成講座 スタンダードコース 2 自己紹介 1995 年 3 月 高知工業高等専門学校
バイオインフォマティクス次世代シーケンサー (NGS) 編 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 バイオインフォマティクス人材育成講座 スタンダードコース 2 自己紹介 1995 年 3 月 高知工業高等専門学校
特論I
 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 2 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG 概論 現状や展望など 講義のみ 第 2 回 (2014
講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 2 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG 概論 現状や展望など 講義のみ 第 2 回 (2014
基本的な利用法
 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握 ) 4. ありがちなミス のところで示しているエラーメッセージとその原因をきっちり理解
到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握 ) 4. ありがちなミス のところで示しているエラーメッセージとその原因をきっちり理解
IonTorrent RNA-Seq 解析概要 サーモフィッシャーサイエンティフィックライフテクノロジーズジャパンテクニカルサポート The world leader in serving science
 IonTorrent RNA-Seq 解析概要 2017-03 サーモフィッシャーサイエンティフィックライフテクノロジーズジャパンテクニカルサポート The world leader in serving science 資料概要 この資料は IonTorrent シーケンサーで RNA-Seq (WholeTranscriptome mrna ampliseqrna mirna) 解析を実施されるユーザー様向けの内容となっています
IonTorrent RNA-Seq 解析概要 2017-03 サーモフィッシャーサイエンティフィックライフテクノロジーズジャパンテクニカルサポート The world leader in serving science 資料概要 この資料は IonTorrent シーケンサーで RNA-Seq (WholeTranscriptome mrna ampliseqrna mirna) 解析を実施されるユーザー様向けの内容となっています
GWB
 NGS データ解析入門 Web セミナー : De Novo シークエンス解析編 1 NGS 新規ゲノム配列解析の手順 シークエンス 遺伝子領域の検出 アセンブル データベース検索 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 前処理 コンティグ配列の作成 CLC Genomics Workbench 遺伝子領域の検出 Blast2GO PRO データベース検索
NGS データ解析入門 Web セミナー : De Novo シークエンス解析編 1 NGS 新規ゲノム配列解析の手順 シークエンス 遺伝子領域の検出 アセンブル データベース検索 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 前処理 コンティグ配列の作成 CLC Genomics Workbench 遺伝子領域の検出 Blast2GO PRO データベース検索
NGSハンズオン講習会
 207.08.08 版 プラスアルファの内容です NGS 解析 ( 初 ~ 中級 ) ゲノムアセンブリ後の各種解析の補足資料 ( プラスアルファ ) 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム寺田朋子 門田幸二 Aug 29-30 207 Contents Gepard でドットプロット 連載第 8 回 W5-3 で最も長い sequence 同士のドットプロットを実行できなかったが
207.08.08 版 プラスアルファの内容です NGS 解析 ( 初 ~ 中級 ) ゲノムアセンブリ後の各種解析の補足資料 ( プラスアルファ ) 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム寺田朋子 門田幸二 Aug 29-30 207 Contents Gepard でドットプロット 連載第 8 回 W5-3 で最も長い sequence 同士のドットプロットを実行できなかったが
機能ゲノム学(第6回)
") RNAseqによる 定 量 的 解 析 とqPCR マイクロアレイなど との 比 較 東 京 大 学 大 学 院 農 学 生 命 科 学 研 究 科 アグリバイオインフォマティクス 教 育 研 究 ユニット 門 田 幸 二 (かどた こうじ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 自 己 紹 介 1995
RNAseqによる 定 量 的 解 析 とqPCR マイクロアレイなど との 比 較 東 京 大 学 大 学 院 農 学 生 命 科 学 研 究 科 アグリバイオインフォマティクス 教 育 研 究 ユニット 門 田 幸 二 (かどた こうじ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ kadota@iu.a.u-tokyo.ac.jp 1 自 己 紹 介 1995
機能ゲノム学
 08.05.08 版 講義資料 PDF が講義のページからダウンロード可能です 講義資料の印刷物はありません 課題用の A4 一枚はあります 第 回出席予定の持込み PC の方は 当日までに Java のインストールをしておいてください 機能ゲノム学第 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム 微生物科学イノベーション連携研究機構門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
08.05.08 版 講義資料 PDF が講義のページからダウンロード可能です 講義資料の印刷物はありません 課題用の A4 一枚はあります 第 回出席予定の持込み PC の方は 当日までに Java のインストールをしておいてください 機能ゲノム学第 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム 微生物科学イノベーション連携研究機構門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
農学生命情報科学特論I
 2015.07.01 版 USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 前回 (6/23) の hoge フォルダがデスクトップに残っているかもしれないのでご注意ください 農学生命情報科学 特論 I 第 3 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
2015.07.01 版 USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 前回 (6/23) の hoge フォルダがデスクトップに残っているかもしれないのでご注意ください 農学生命情報科学 特論 I 第 3 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
リード・ゲノム・アノテーションインポート
 リード ゲノム アノテーションインポート 1 Location と Folder ロケーション フォルダ Genomics Workbenchではデータを以下のような階層構造で保存可能です フォルダの一番上位の階層を Location と呼び その下の階層を Folder と呼びます データの保存場所はロケーション毎に設定可能です たとえばあるデータは C ドライブに保存し あるデータは D ドライブに保存するといった事が可能です
リード ゲノム アノテーションインポート 1 Location と Folder ロケーション フォルダ Genomics Workbenchではデータを以下のような階層構造で保存可能です フォルダの一番上位の階層を Location と呼び その下の階層を Folder と呼びます データの保存場所はロケーション毎に設定可能です たとえばあるデータは C ドライブに保存し あるデータは D ドライブに保存するといった事が可能です
PowerPoint プレゼンテーション
 V1 次世代シークエンサ実習 II 本講義の内容 Reseq 解析 RNA-seq 解析 公開データ取得 クオリティコントロール マッピング 変異検出 公開データ取得 クオリティコントロール マッピング 発現定量 FPKM を算出します 2 R N A - s e q とは メッセンジャー RNA(mRNA) をキャプチャして次世代シーケンサーでシーケンシングする手法 リファレンスがある生物種の場合
V1 次世代シークエンサ実習 II 本講義の内容 Reseq 解析 RNA-seq 解析 公開データ取得 クオリティコントロール マッピング 変異検出 公開データ取得 クオリティコントロール マッピング 発現定量 FPKM を算出します 2 R N A - s e q とは メッセンジャー RNA(mRNA) をキャプチャして次世代シーケンサーでシーケンシングする手法 リファレンスがある生物種の場合
1. はじめに 1. はじめに 1-1. KaPPA-Average とは KaPPA-Average は KaPPA-View( でマイクロアレイデータを解析する際に便利なデータ変換ソフトウェアです 一般のマイクロアレイでは 一つのプロー
 KaPPA-Average 1.0 マニュアル 第 1.0 版 制作者 : かずさ DNA 研究所櫻井望 制作日 : 2010 年 1 月 12 日 目次 1. はじめに 2 1-1. KaPPA-Average とは 2 1-2. 動作環境 3 1-3. インストールと起動 3 2. 操作説明 4 2-1. メイン機能 - Calc. Average 4 2-1-1. データの準備 4 2-1-2.
KaPPA-Average 1.0 マニュアル 第 1.0 版 制作者 : かずさ DNA 研究所櫻井望 制作日 : 2010 年 1 月 12 日 目次 1. はじめに 2 1-1. KaPPA-Average とは 2 1-2. 動作環境 3 1-3. インストールと起動 3 2. 操作説明 4 2-1. メイン機能 - Calc. Average 4 2-1-1. データの準備 4 2-1-2.
ゲノム情報解析基礎 ~ Rで塩基配列解析 ~
 トランスクリプトーム解析の現況 ~ マイクロアレイ vs. RNA-seq~ 東京大学 大学院農学生命科学研究科 アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 スライド PDF はウェブから取得可能です 2 ステレオタイプなイメージ
トランスクリプトーム解析の現況 ~ マイクロアレイ vs. RNA-seq~ 東京大学 大学院農学生命科学研究科 アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 スライド PDF はウェブから取得可能です 2 ステレオタイプなイメージ
nagasaki_GMT2015_key09
 Workflow Variant Calling 03 長崎は遺伝研 大量遺伝情報研究室の所属です 国立遺伝学研究所 生命情報研究センター 3F 2F 欧州EBIと米国NCBIと密接に協力しながら DDBJ/EMBL/GenBank国際塩基配列データ ベースを構築しています 私たちは 塩基配列登録を支援するシステムづくり 登録データを活用するシステムづくり 高速シーケンス配列の情報解析 を行なっています
Workflow Variant Calling 03 長崎は遺伝研 大量遺伝情報研究室の所属です 国立遺伝学研究所 生命情報研究センター 3F 2F 欧州EBIと米国NCBIと密接に協力しながら DDBJ/EMBL/GenBank国際塩基配列データ ベースを構築しています 私たちは 塩基配列登録を支援するシステムづくり 登録データを活用するシステムづくり 高速シーケンス配列の情報解析 を行なっています
GWB
 NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 CLC Genomics Workbench 使用ツール シークエンスデータのインポート NGS data import クオリティチェック QC for Sequencing Reads Trim Reads 参照ゲノム配列へのマッピング 再アライメント
NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 CLC Genomics Workbench 使用ツール シークエンスデータのインポート NGS data import クオリティチェック QC for Sequencing Reads Trim Reads 参照ゲノム配列へのマッピング 再アライメント
GWB_RNA-Seq_
 CLC Genomics Workbench ウェブトレーニングセミナー : RNA-Seq 編 フィルジェン株式会社バイオサイエンス部 (biosupport@filgen.jp) 1 Advanced RNA-Seq プラグイン CLC Genomics Workbench 9.0 / Biomedical Genomics Workbench 3.0 以降で使用可能な無償プラグイン RNA-Seq
CLC Genomics Workbench ウェブトレーニングセミナー : RNA-Seq 編 フィルジェン株式会社バイオサイエンス部 (biosupport@filgen.jp) 1 Advanced RNA-Seq プラグイン CLC Genomics Workbench 9.0 / Biomedical Genomics Workbench 3.0 以降で使用可能な無償プラグイン RNA-Seq
RNA-seq
 RNA-seq 1 RNA-seq 解析フロー RNA-seq インポート クオリティチェック RNA-seq 発現差解析 この資料では RNA-seq からの説明となりますが インポート クオリティチェックについては サポート資料のページより内容をご確認いただけます 2 データ 発現解析用デモデータは 以下よりダウンロードいただけます ES 細胞 (ESC) と神経前駆細胞 (NPC) の発現解析を小さなデモデータで行えます
RNA-seq 1 RNA-seq 解析フロー RNA-seq インポート クオリティチェック RNA-seq 発現差解析 この資料では RNA-seq からの説明となりますが インポート クオリティチェックについては サポート資料のページより内容をご確認いただけます 2 データ 発現解析用デモデータは 以下よりダウンロードいただけます ES 細胞 (ESC) と神経前駆細胞 (NPC) の発現解析を小さなデモデータで行えます
ゲノム情報解析基礎
 ゲノム情報解析基礎 ~ R で塩基配列解析 ~ 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 多くのヒトが感想を述べられて 感想やコメント へのコメントいました ありがとうございます コピペではなく位置から自分が入力するのは無理そう
ゲノム情報解析基礎 ~ R で塩基配列解析 ~ 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 多くのヒトが感想を述べられて 感想やコメント へのコメントいました ありがとうございます コピペではなく位置から自分が入力するのは無理そう
Rでゲノム・トランスクリプトーム解析
 06.03.05 版 実習用 PC のデスクトップ上に hoge フォルダがあります この中に解析に必要な入力ファイルがあります ネットワーク不具合時は ローカル環境で html ファイルを起動して各自対応してください R で塩基配列解析 : ゲノム解析からトランスクリプトーム解析まで 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ )
06.03.05 版 実習用 PC のデスクトップ上に hoge フォルダがあります この中に解析に必要な入力ファイルがあります ネットワーク不具合時は ローカル環境で html ファイルを起動して各自対応してください R で塩基配列解析 : ゲノム解析からトランスクリプトーム解析まで 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ )
基本的な利用法
 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握 ) 4. ありがちなミス のところで示しているエラーメッセージとその原因をきっちり理解
到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握 ) 4. ありがちなミス のところで示しているエラーメッセージとその原因をきっちり理解
PrimerArray® Analysis Tool Ver.2.2
 研究用 PrimerArray Analysis Tool Ver.2.2 説明書 v201801 PrimerArray Analysis Tool Ver.2.2 は PrimerArray( 製品コード PH001 ~ PH007 PH009 ~ PH015 PN001 ~ PN015) で得られたデータを解析するためのツールで コントロールサンプルと 1 種類の未知サンプル間の比較が可能です
研究用 PrimerArray Analysis Tool Ver.2.2 説明書 v201801 PrimerArray Analysis Tool Ver.2.2 は PrimerArray( 製品コード PH001 ~ PH007 PH009 ~ PH015 PN001 ~ PN015) で得られたデータを解析するためのツールで コントロールサンプルと 1 種類の未知サンプル間の比較が可能です
ゲノム情報解析基礎
 講義資料 PDF が講義のページからダウンロード可能です 印刷物はありません ゲノム情報解析基礎 ~ R で塩基配列解析 ~ 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム 微生物科学イノベーション連携研究機構門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 講義予定
講義資料 PDF が講義のページからダウンロード可能です 印刷物はありません ゲノム情報解析基礎 ~ R で塩基配列解析 ~ 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム 微生物科学イノベーション連携研究機構門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 講義予定
特論I
 2016.02.01 版 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 3 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp Jun 25, 2014 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG
2016.02.01 版 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 3 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 kadota@iu.a.u-tokyo.ac.jp Jun 25, 2014 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG
機能ゲノム学(第6回)
") トランスクリプトーム 解析手法の開発 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス人材養成ユニット門田幸二 2008/12/08 トランスクリプトーム (transcrptome) とは 細胞中に存在する転写物全体 (transcrpt + ome) トランスクリプトーム解析技術 DNA マイクロアレイ Affymetrx GeneChp, cdna アレイ, 電気泳動に基づく方法
トランスクリプトーム 解析手法の開発 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス人材養成ユニット門田幸二 2008/12/08 トランスクリプトーム (transcrptome) とは 細胞中に存在する転写物全体 (transcrpt + ome) トランスクリプトーム解析技術 DNA マイクロアレイ Affymetrx GeneChp, cdna アレイ, 電気泳動に基づく方法
ChIP-seq
 ChIP-seq 1 ChIP-seq 解析原理 ChIP サンプルのフラグメントでは タンパク質結合部位付近にそれぞれ Forward と Reverse のリードがマップされることが予想される ChIP のサンプルでは Forward と Reverse のリードを 3 側へシフトさせ ChIP のピークを算出する コントロールサンプルでは ChIP のサンプルとは異なり 特定の場所に多くマップされないため
ChIP-seq 1 ChIP-seq 解析原理 ChIP サンプルのフラグメントでは タンパク質結合部位付近にそれぞれ Forward と Reverse のリードがマップされることが予想される ChIP のサンプルでは Forward と Reverse のリードを 3 側へシフトさせ ChIP のピークを算出する コントロールサンプルでは ChIP のサンプルとは異なり 特定の場所に多くマップされないため
Qlucore_seminar_slide_180604
 シングルセル RNA-Seq のための 情報解析 フィルジェン株式会社バイオサイエンス部 (biosupport@filgen.jp) 1 シングルセル RNA-Seq シングルセル RNA-Seq のデータ解析では 通常の RNA-Seq データの解析手法に加え データセット内の各細胞の遺伝子発現プロファイルの違いを俯瞰できるような 強力な情報解析アルゴリズムと データのビジュアライズ機能を利用する必要がある
シングルセル RNA-Seq のための 情報解析 フィルジェン株式会社バイオサイエンス部 (biosupport@filgen.jp) 1 シングルセル RNA-Seq シングルセル RNA-Seq のデータ解析では 通常の RNA-Seq データの解析手法に加え データセット内の各細胞の遺伝子発現プロファイルの違いを俯瞰できるような 強力な情報解析アルゴリズムと データのビジュアライズ機能を利用する必要がある
データ科学2.pptx
 データ科学 多重検定 2 mul%ple test False Discovery Rate 藤博幸 前回の復習 1 多くの検定を繰り返す時には 単純に個々の検定を繰り返すだけでは不十分 5% 有意水準ということは, 1000 回検定を繰り返すと, 50 回くらいは帰無仮説が正しいのに 間違って棄却されてすまうじちがあるということ ex) 1 万個の遺伝子について 正常細胞とガン細胞で それぞれの遺伝子の発現に差があるかどうかを検定
データ科学 多重検定 2 mul%ple test False Discovery Rate 藤博幸 前回の復習 1 多くの検定を繰り返す時には 単純に個々の検定を繰り返すだけでは不十分 5% 有意水準ということは, 1000 回検定を繰り返すと, 50 回くらいは帰無仮説が正しいのに 間違って棄却されてすまうじちがあるということ ex) 1 万個の遺伝子について 正常細胞とガン細胞で それぞれの遺伝子の発現に差があるかどうかを検定
NGS速習コース
 バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-4. R Bioconductor I 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-4. R Bioconductor I 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
KEGG.ppt
 1 2 3 4 KEGG: Kyoto Encyclopedia of Genes and Genomes http://www.genome.jp/kegg/kegg2.html http://www.genome.jp/kegg/kegg_ja.html 5 KEGG PATHWAY 生体内(外)の分子間ネットワーク図 代謝系 12カテゴリ 中間代謝 二次代謝 薬の 代謝 全体像 制御系 20カテゴリ
1 2 3 4 KEGG: Kyoto Encyclopedia of Genes and Genomes http://www.genome.jp/kegg/kegg2.html http://www.genome.jp/kegg/kegg_ja.html 5 KEGG PATHWAY 生体内(外)の分子間ネットワーク図 代謝系 12カテゴリ 中間代謝 二次代謝 薬の 代謝 全体像 制御系 20カテゴリ
Rインストール手順
 R 本体は最新のリリース版 R パッケージは 必要最小限プラスアルファ の推奨インストール手順を示します スライドは Windows0 環境でのスクリーンショットです ウェブブラウザによって挙動が多少異なるのでご注意ください 私は Chrome を使っています R 本体とパッケージのインストール Windows 版 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二
R 本体は最新のリリース版 R パッケージは 必要最小限プラスアルファ の推奨インストール手順を示します スライドは Windows0 環境でのスクリーンショットです ウェブブラウザによって挙動が多少異なるのでご注意ください 私は Chrome を使っています R 本体とパッケージのインストール Windows 版 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二
 UCSC ゲノムブラウザチュートリアル UCSC ゲノムブラウザはゲノム解読がなされている真核生物を対象として自動アノテーションを行い その結果をデータベースとして公開している UCSC が進めているプロジェクトです NCBI MapViewer のようにゲノムベースでその上にアノテーションされている遺伝子などの情報を閲覧すると共に ホモロジー検索や必要なデータのダウンロードなどの機能を提供しています
UCSC ゲノムブラウザチュートリアル UCSC ゲノムブラウザはゲノム解読がなされている真核生物を対象として自動アノテーションを行い その結果をデータベースとして公開している UCSC が進めているプロジェクトです NCBI MapViewer のようにゲノムベースでその上にアノテーションされている遺伝子などの情報を閲覧すると共に ホモロジー検索や必要なデータのダウンロードなどの機能を提供しています
GWB
 NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 参照ゲノム配列へのマッピング 再アライメント 変異検出 CLC Genomics Workbench または Biomedical Genomics Workbench
NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 参照ゲノム配列へのマッピング 再アライメント 変異検出 CLC Genomics Workbench または Biomedical Genomics Workbench
Microsoft Word - 1 color Normalization Document _Agilent version_ .doc
 color 実験の Normalization color 実験で得られた複数のアレイデータを相互比較するためには Normalization( 正規化 ) が必要です 2 つのサンプルを異なる色素でラベル化し 競合ハイブリダイゼーションさせる 2color 実験では 基本的に Dye Normalization( 色素補正 ) が適用されますが color 実験では データの特徴と実験の目的 (
color 実験の Normalization color 実験で得られた複数のアレイデータを相互比較するためには Normalization( 正規化 ) が必要です 2 つのサンプルを異なる色素でラベル化し 競合ハイブリダイゼーションさせる 2color 実験では 基本的に Dye Normalization( 色素補正 ) が適用されますが color 実験では データの特徴と実験の目的 (
CLC Genomics Workbench ウェブトレーニングセミナー: 変異解析編
 CLC Genomics Workbench ウェブトレーニングセミナー : 遺伝子発現解析編 12 th Feb., 2016 フィルジェン株式会社バイオサイエンス部 biosupport@filgen.jp Feb., 2016_V2 1 遺伝子発現解析概要 本日のセミナーにおける解析の流れ及び使用するツール名 ( 図中赤枠部分 ) Case Control インポート インポート インポート
CLC Genomics Workbench ウェブトレーニングセミナー : 遺伝子発現解析編 12 th Feb., 2016 フィルジェン株式会社バイオサイエンス部 biosupport@filgen.jp Feb., 2016_V2 1 遺伝子発現解析概要 本日のセミナーにおける解析の流れ及び使用するツール名 ( 図中赤枠部分 ) Case Control インポート インポート インポート
講義内容 ファイル形式 データの可視化 データのクオリティチェック マッピング アセンブル 資料の見方 $ pwd 実際に入力するコマンドを黄色い四角の中に示します 2
 N G S 解析基礎 講義内容 ファイル形式 データの可視化 データのクオリティチェック マッピング アセンブル 資料の見方 $ pwd 実際に入力するコマンドを黄色い四角の中に示します 2 ファイル形式 NGS 解析でよく使われるファイル形式 ファイル形式 fastq bam/sam vcf bed fasta サンプルデータの場所 /home/ ユーザ名 /Desktop/amelieff/1K_ERR038793_1.fastq
N G S 解析基礎 講義内容 ファイル形式 データの可視化 データのクオリティチェック マッピング アセンブル 資料の見方 $ pwd 実際に入力するコマンドを黄色い四角の中に示します 2 ファイル形式 NGS 解析でよく使われるファイル形式 ファイル形式 fastq bam/sam vcf bed fasta サンプルデータの場所 /home/ ユーザ名 /Desktop/amelieff/1K_ERR038793_1.fastq
PowerPoint Presentation
 エピジェノミクス解析編 2016/08/10 Filgen ChIP-seq (Transfactor & Histone), Bisulfite webex seminar 株式会社キアゲンアプライドアドバンストゲノミクス宮本真理, PhD 1 アジェンダ ChIP-seq 解析 Transcription Factor ChIP-seq Histone ChIP-seq Bisulfite-seq
エピジェノミクス解析編 2016/08/10 Filgen ChIP-seq (Transfactor & Histone), Bisulfite webex seminar 株式会社キアゲンアプライドアドバンストゲノミクス宮本真理, PhD 1 アジェンダ ChIP-seq 解析 Transcription Factor ChIP-seq Histone ChIP-seq Bisulfite-seq
RNA-seq
 CLC Genomics Workbench ハンズオントレーニング RNA-seq 株式会社 CLCバイオジャパンシニアフィールドバイオインフォマティクスサイエンティスト宮本真理 Ph.D. mmiyamoto@clcbio.co.jp 1 support@clcbio.co.jp 2 アジェンダ Genomics Workbench 概要 今日のデータ RNA-seq 解析 データインポート QC
CLC Genomics Workbench ハンズオントレーニング RNA-seq 株式会社 CLCバイオジャパンシニアフィールドバイオインフォマティクスサイエンティスト宮本真理 Ph.D. mmiyamoto@clcbio.co.jp 1 support@clcbio.co.jp 2 アジェンダ Genomics Workbench 概要 今日のデータ RNA-seq 解析 データインポート QC
NGSハンズオン講習会
 205.07.27 版 配布する USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください NGS ハンズオン 講習会 :R 基礎 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ Contents(
205.07.27 版 配布する USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください NGS ハンズオン 講習会 :R 基礎 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp http://www.iu.a.u-tokyo.ac.jp/~kadota/ Contents(
141025mishima
 NGS (RNAseq) »NGS Now Generation Sequencer»NGS»» 4 NGS(Next Generation Sequencer) Now Generation Sequencer http://www.youtube.com/watch?v=womkfikwlxm http://www.youtube.com/watch?v=mxkya9xcvbq http://www.youtube.com/watch?v=nhcj8ptycfc
NGS (RNAseq) »NGS Now Generation Sequencer»NGS»» 4 NGS(Next Generation Sequencer) Now Generation Sequencer http://www.youtube.com/watch?v=womkfikwlxm http://www.youtube.com/watch?v=mxkya9xcvbq http://www.youtube.com/watch?v=nhcj8ptycfc
V1 ゲノム R e s e q 変異解析 Copyright Amelieff Corporation All Rights Reserved.
 V1 ゲノム R e s e q 変異解析 本講義にあたって 代表的な解析の流れを紹介します 論文でよく使用されているツールを使用します コマンドを沢山実行します スペルミスが心配な方は コマンド例がありますのでコピーし て実行してください マークのコマンドは実行してください 実行が遅れてもあせらずに 応用や課題の間に追い付いてくだ さい 2 本講義の内容 Reseq解析 RNA-seq解析 公開データ取得
V1 ゲノム R e s e q 変異解析 本講義にあたって 代表的な解析の流れを紹介します 論文でよく使用されているツールを使用します コマンドを沢山実行します スペルミスが心配な方は コマンド例がありますのでコピーし て実行してください マークのコマンドは実行してください 実行が遅れてもあせらずに 応用や課題の間に追い付いてくだ さい 2 本講義の内容 Reseq解析 RNA-seq解析 公開データ取得
れており 世界的にも重要課題とされています それらの中で 非常に高い完全長 cdna のカバー率を誇るマウスエンサイクロペディア計画は極めて重要です ゲノム科学総合研究センター (GSC) 遺伝子構造 機能研究グループでは これまでマウス完全長 cdna100 万クローン以上の末端塩基配列データを
 遺伝子構造 機能研究グループでは これまでマウス完全長 cdna100 万クローン以上の末端塩基配列データを") 報道発表資料 2002 年 12 月 5 日 独立行政法人理化学研究所 遺伝子の機能解析を飛躍的に進める世界最大規模の遺伝子情報を公開 - 遺伝子として認知されていなかった部分が転写されていることを実証 - 理化学研究所 ( 小林俊一理事長 ) は マウスの完全長 cdna 160,770 クローンの塩基配列および機能アノテーション ( 機能注釈 ) 情報を公開します これは 現在までに人類が収得している遺伝子の約
報道発表資料 2002 年 12 月 5 日 独立行政法人理化学研究所 遺伝子の機能解析を飛躍的に進める世界最大規模の遺伝子情報を公開 - 遺伝子として認知されていなかった部分が転写されていることを実証 - 理化学研究所 ( 小林俊一理事長 ) は マウスの完全長 cdna 160,770 クローンの塩基配列および機能アノテーション ( 機能注釈 ) 情報を公開します これは 現在までに人類が収得している遺伝子の約
Microsoft PowerPoint - 6_TS-0891(TS-0835(Custom TaqMan Assay Design Tool利用方法修正5.pptx
 Custom TaqMan Assay Design Tool インターネットオーダー方法 20010/06/01 Custom TaqMan Assay Design Tool 1. Custom TaqMan SNP Genotyping Assays P.5 2. Custom TaqMan Gene Expression Assays P.21 3. カスタムデザインでのオーダー P.30
Custom TaqMan Assay Design Tool インターネットオーダー方法 20010/06/01 Custom TaqMan Assay Design Tool 1. Custom TaqMan SNP Genotyping Assays P.5 2. Custom TaqMan Gene Expression Assays P.21 3. カスタムデザインでのオーダー P.30
PowerPoint プレゼンテーション
 V1 次世代シークエンサ実習 II 本講義にあたって 代表的な解析の流れを紹介します 論文でよく使用されているツールを使用します コマンドを沢山実行します スペルミスが心配な方は コマンド例がありますのでコピーして実行してください /home/admin1409/amelieff/ngs/reseq_command.txt マークのコマンドは実行してください 実行が遅れてもあせらずに 応用や課題の間に追い付いてください
V1 次世代シークエンサ実習 II 本講義にあたって 代表的な解析の流れを紹介します 論文でよく使用されているツールを使用します コマンドを沢山実行します スペルミスが心配な方は コマンド例がありますのでコピーして実行してください /home/admin1409/amelieff/ngs/reseq_command.txt マークのコマンドは実行してください 実行が遅れてもあせらずに 応用や課題の間に追い付いてください
Apache-Tomcat と 冗長な UTF-8 表現 (CVE 検証レポート ) 2008 年 08 月 26 日 Ver. 0.1
 2008 年 08 月 26 日 Ver. 0.1") Apache-Tomcat と 冗長な UTF-8 表現 (CVE-2008-2938 検証レポート ) 2008 年 08 月 26 日 Ver. 0.1 目次 1 調査概要 2 2 UTF-8 とは 3 3 CVE-208-2938 4 3.1.( ピリオド ) について 4 4 CVE-208-2938 と3Byteの冗長な UTF-8 表現 5 5 CVE-208-2938 と /( スラッシュ
Apache-Tomcat と 冗長な UTF-8 表現 (CVE-2008-2938 検証レポート ) 2008 年 08 月 26 日 Ver. 0.1 目次 1 調査概要 2 2 UTF-8 とは 3 3 CVE-208-2938 4 3.1.( ピリオド ) について 4 4 CVE-208-2938 と3Byteの冗長な UTF-8 表現 5 5 CVE-208-2938 と /( スラッシュ
AJACS18_ ppt
 1, 1, 1, 1, 1, 1,2, 1,2, 1 1 DDBJ 2 AJACS3 2010 6 414:20-15:20 2231 DDBJ DDBJ DDBJ DDBJ NCBI (GenBank) DDBJ EBI (EMBL-Bank) GEO DDBJ Omics ARchive(DOR) ArrayExpress DTA (DDBJ Trace Archive) DRA (DDBJ
1, 1, 1, 1, 1, 1,2, 1,2, 1 1 DDBJ 2 AJACS3 2010 6 414:20-15:20 2231 DDBJ DDBJ DDBJ DDBJ NCBI (GenBank) DDBJ EBI (EMBL-Bank) GEO DDBJ Omics ARchive(DOR) ArrayExpress DTA (DDBJ Trace Archive) DRA (DDBJ
Maser - User Operation Manual
 Maser 3 Cell Innovation User Operation Manual 2013.4.1 1 目次 1. はじめに... 3 1.1. 推奨動作環境... 3 2. データの登録... 4 2.1. プロジェクトの作成... 4 2.2. Projectへのデータのアップロード... 8 2.2.1. HTTPSでのアップロード... 8 2.2.2. SFTPでのアップロード...
Maser 3 Cell Innovation User Operation Manual 2013.4.1 1 目次 1. はじめに... 3 1.1. 推奨動作環境... 3 2. データの登録... 4 2.1. プロジェクトの作成... 4 2.2. Projectへのデータのアップロード... 8 2.2.1. HTTPSでのアップロード... 8 2.2.2. SFTPでのアップロード...
 GenBank クイックスタート GenBank は NLM/NCBI にて維持管理されている核酸配列データベースです また GenBank は EMBL, DDBJ と三極間で連携しながら国際核酸配列データベースを共同で構築しています これら三機関はデータを日々交換し続けており その規模は 160000 種にも及ぶ生物種の塩基配列から成り立つまでになっています この GenBank クイックスタートでは
GenBank クイックスタート GenBank は NLM/NCBI にて維持管理されている核酸配列データベースです また GenBank は EMBL, DDBJ と三極間で連携しながら国際核酸配列データベースを共同で構築しています これら三機関はデータを日々交換し続けており その規模は 160000 種にも及ぶ生物種の塩基配列から成り立つまでになっています この GenBank クイックスタートでは
Microsoft PowerPoint - install_NGSsokushu_windows(ver2.1).pptx
.pptx") 2014/08/22 作成 (ver. 2.1) VirtualBox のインストールと 設定済み BioLinux7 の導入 (BioLinux7.ova) Windows 7 (64bit) 環境に.ova ファイル形式の BioLinux7 ( 配布中 ) を導入する場合 注意事項 ) 実習では.ova ファイルから BioLinux7 の導入を行って下さい VirtualBox は最新版ではなく
2014/08/22 作成 (ver. 2.1) VirtualBox のインストールと 設定済み BioLinux7 の導入 (BioLinux7.ova) Windows 7 (64bit) 環境に.ova ファイル形式の BioLinux7 ( 配布中 ) を導入する場合 注意事項 ) 実習では.ova ファイルから BioLinux7 の導入を行って下さい VirtualBox は最新版ではなく
農業・農村基盤図の大字小字コードXML作成 説明書
 農業 農村基盤図の大字小字コード XML 作成説明書 2007/06/06 有限会社ジオ コーチ システムズ http://www.geocoach.co.jp/ info@geocoach.co.jp 農業 農村基盤図の大字小字コード XML 作成 プログラムについての説明書です バージョン ビルド 1.01 2007/06/06 農業 農村基盤図の大字小字コード XML 作成 は 市区町村 大字
農業 農村基盤図の大字小字コード XML 作成説明書 2007/06/06 有限会社ジオ コーチ システムズ http://www.geocoach.co.jp/ info@geocoach.co.jp 農業 農村基盤図の大字小字コード XML 作成 プログラムについての説明書です バージョン ビルド 1.01 2007/06/06 農業 農村基盤図の大字小字コード XML 作成 は 市区町村 大字
2016_RNAseq解析_修正版
 平成 28 年度 NGS ハンズオン講習会 RNA-seq 解析 2016 年 7 27 本講義にあたって n 代表的な解析の流れを紹介します 論 でよく使 されているツールを使 します n コマンドを沢 実 します タイプミスが 配な は コマンド例がありますのでコピーして実 してください 実 が遅れてもあせらずに 課題や休憩の間に追い付いてください Amelieff Corporation All
平成 28 年度 NGS ハンズオン講習会 RNA-seq 解析 2016 年 7 27 本講義にあたって n 代表的な解析の流れを紹介します 論 でよく使 されているツールを使 します n コマンドを沢 実 します タイプミスが 配な は コマンド例がありますのでコピーして実 してください 実 が遅れてもあせらずに 課題や休憩の間に追い付いてください Amelieff Corporation All
AJACS_komachi.key
 Tweet OK 統合データベース講習会 AJACSこまち 塩基配列解析のための データベース ウェブツールと CRISPRガイドRNA設計 ライフサイエンス統合データベースセンター (DBCLS) 内藤雄樹 自己紹介 内藤 雄樹 ないとう ゆうき @meso_cacase ライフサイエンス統合データベース センター DBCLS 特任助教 過去に RNAi メカニズム等の研究 sirna設計サイト:
Tweet OK 統合データベース講習会 AJACSこまち 塩基配列解析のための データベース ウェブツールと CRISPRガイドRNA設計 ライフサイエンス統合データベースセンター (DBCLS) 内藤雄樹 自己紹介 内藤 雄樹 ないとう ゆうき @meso_cacase ライフサイエンス統合データベース センター DBCLS 特任助教 過去に RNAi メカニズム等の研究 sirna設計サイト:
KEGG_PATHWAY.ppt
 KEGG: Kyoto Encyclopedia of Genes and Genomes KEGG: Kyoto Encyclopedia of Genes and Genomes 様々な種類のデータを 生命現象の総体 として再構築 ツールの提供 EGassembler KAAS GENIES ネットワークの知識 ツールの提供 e-zyme pathcomp 高次機能 機能の階層分類 相互参照用データ
KEGG: Kyoto Encyclopedia of Genes and Genomes KEGG: Kyoto Encyclopedia of Genes and Genomes 様々な種類のデータを 生命現象の総体 として再構築 ツールの提供 EGassembler KAAS GENIES ネットワークの知識 ツールの提供 e-zyme pathcomp 高次機能 機能の階層分類 相互参照用データ
ThermoFisher
 Thermo Fisher Connect Relative Quantification 操作簡易資料 http://www.thermofisher.com/cloud 使用には事前登録が必要になります 画面は予告なく変わることがあります The world leader in serving science Thermo Fisher Connect とは? キャピラリシーケンサ リアルタイム
Thermo Fisher Connect Relative Quantification 操作簡易資料 http://www.thermofisher.com/cloud 使用には事前登録が必要になります 画面は予告なく変わることがあります The world leader in serving science Thermo Fisher Connect とは? キャピラリシーケンサ リアルタイム
win版8日目
 8 日目 : 項目のチェック (2) 1 日 30 分くらい,30 日で何とか R をそこそこ使えるようになるための練習帳 :Win 版 昨日は, 平均値などの基礎統計量を計算する試行錯誤へご招待しましたが (?), 今日は簡 単にやってみます そのためには,psych というパッケージが必要となりますが, パッケー ジのインストール & 読み込みの詳しい方法は, 後で説明します 以下の説明は,psych
8 日目 : 項目のチェック (2) 1 日 30 分くらい,30 日で何とか R をそこそこ使えるようになるための練習帳 :Win 版 昨日は, 平均値などの基礎統計量を計算する試行錯誤へご招待しましたが (?), 今日は簡 単にやってみます そのためには,psych というパッケージが必要となりますが, パッケー ジのインストール & 読み込みの詳しい方法は, 後で説明します 以下の説明は,psych
Microsoft Word - index.html
 R のインストールと超入門 R とは? R ダウンロード R のインストール R の基本操作 R 終了データの読み込みパッケージの操作 R とは? Rはデータ解析 マイニングを行うフリーソフトである Rはデータ解析の環境でもあり 言語でもある ニュージーランドのオークランド (Auckland) 大学の統計学科のRobert Gentlemanと Ross Ihakaにより開発がはじめられ 1997
R のインストールと超入門 R とは? R ダウンロード R のインストール R の基本操作 R 終了データの読み込みパッケージの操作 R とは? Rはデータ解析 マイニングを行うフリーソフトである Rはデータ解析の環境でもあり 言語でもある ニュージーランドのオークランド (Auckland) 大学の統計学科のRobert Gentlemanと Ross Ihakaにより開発がはじめられ 1997
国際塩基配列データベース n DNA のデータベース GenBank ( アメリカ :Na,onal Center for Biotechnology Informa,on, NCBI が運営 ) EMBL ( ヨーロッパ : 欧州生命情報学研究所が運営 ) DDBJ ( 日本 : 国立遺伝研内の日
 EMBL ( ヨーロッパ : 欧州生命情報学研究所が運営 ) DDBJ ( 日本 : 国立遺伝研内の日") 生物情報工学 BioInforma*cs 3 遺伝子データベース 16/06/09 1 国際塩基配列データベース n DNA のデータベース GenBank ( アメリカ :Na,onal Center for Biotechnology Informa,on, NCBI が運営 ) EMBL ( ヨーロッパ : 欧州生命情報学研究所が運営 ) DDBJ ( 日本 : 国立遺伝研内の日本 DNA データバンクが運営
生物情報工学 BioInforma*cs 3 遺伝子データベース 16/06/09 1 国際塩基配列データベース n DNA のデータベース GenBank ( アメリカ :Na,onal Center for Biotechnology Informa,on, NCBI が運営 ) EMBL ( ヨーロッパ : 欧州生命情報学研究所が運営 ) DDBJ ( 日本 : 国立遺伝研内の日本 DNA データバンクが運営
数量的アプローチ 年 6 月 11 日 イントロダクション データ分析をマスターする 12 のレッスン ウェブサポートページ ( 有斐閣 ) 水落研究室 R http:
 水落研究室 R http:") イントロダクション データ分析をマスターする 12 のレッスン ウェブサポートページ ( 有斐閣 ) http://yuhikaku-nibu.txt-nifty.com/blog/2017/09/22103.html 水落研究室 R http://depts.nanzan-u.ac.jp/ugrad/ps/mizuochi/r.html 1 この授業では統計ソフト R を使って分析を行います データを扱うソフトとして
イントロダクション データ分析をマスターする 12 のレッスン ウェブサポートページ ( 有斐閣 ) http://yuhikaku-nibu.txt-nifty.com/blog/2017/09/22103.html 水落研究室 R http://depts.nanzan-u.ac.jp/ugrad/ps/mizuochi/r.html 1 この授業では統計ソフト R を使って分析を行います データを扱うソフトとして
目次 第 1 章はじめに 本ソフトの概要... 2 第 2 章インストール編 ソフトの動作環境を確認しましょう ソフトをコンピュータにセットアップしましょう 動作を確認しましょう コンピュータからアンインストー
 JS 管理ファイル作成支援ソフト 工事用 Ver.4.0 インストールマニュアル 操作マニュアル 日本下水道事業団 目次 第 1 章はじめに... 1 1-1 本ソフトの概要... 2 第 2 章インストール編... 3 2-1 ソフトの動作環境を確認しましょう... 4 2-2 ソフトをコンピュータにセットアップしましょう... 5 2-3 動作を確認しましょう... 8 2-4 コンピュータからアンインストールする方法...
JS 管理ファイル作成支援ソフト 工事用 Ver.4.0 インストールマニュアル 操作マニュアル 日本下水道事業団 目次 第 1 章はじめに... 1 1-1 本ソフトの概要... 2 第 2 章インストール編... 3 2-1 ソフトの動作環境を確認しましょう... 4 2-2 ソフトをコンピュータにセットアップしましょう... 5 2-3 動作を確認しましょう... 8 2-4 コンピュータからアンインストールする方法...
<4D F736F F D208D C8FEE95F18DEC90AC A B D836A B2E646F63>
 国土数値情報作成アプリケーション ( 指定地域データ等生成ツール ) 利用マニュアル 平成 20 年 3 月 国土交通省国土計画局 目次 1. ツール名 1 2. 機能概要 1 3. ツールのインストール 1 4. 使用方法 4 5. 動作環境 10 6. ツールのアンインストール 11 7.FAQ 12 1. ツール名 KSJ 指定地域データ等生成ツール -v#_##.exe (#_## はバージョン番号
国土数値情報作成アプリケーション ( 指定地域データ等生成ツール ) 利用マニュアル 平成 20 年 3 月 国土交通省国土計画局 目次 1. ツール名 1 2. 機能概要 1 3. ツールのインストール 1 4. 使用方法 4 5. 動作環境 10 6. ツールのアンインストール 11 7.FAQ 12 1. ツール名 KSJ 指定地域データ等生成ツール -v#_##.exe (#_## はバージョン番号
Microsoft Word - CygwinでPython.docx
 Cygwin でプログラミング 2018/4/9 千葉 数値計算は計算プログラムを書いて行うわけですが プログラムには様々な 言語 があるので そのうちどれかを選択する必要があります プログラム言語には 人間が書いたプログラムを一度計算機用に翻訳したのち計算を実行するものと 人間が書いたプログラムを計算機が読んでそのまま実行するものとがあります ( 若干不正確な説明ですが ) 前者を システム言語
Cygwin でプログラミング 2018/4/9 千葉 数値計算は計算プログラムを書いて行うわけですが プログラムには様々な 言語 があるので そのうちどれかを選択する必要があります プログラム言語には 人間が書いたプログラムを一度計算機用に翻訳したのち計算を実行するものと 人間が書いたプログラムを計算機が読んでそのまま実行するものとがあります ( 若干不正確な説明ですが ) 前者を システム言語
Rでゲノム・トランスクリプトーム解析
 06.08. 版 スライド 8 までは自習 当日はスライド 9 から始める予定 スライド 3-86 は当日省略予定 講習会後に各自で復習してください 第 3 部 :NGS 解析 ( 中 ~ 上級 ) ~ トランスクリプトームアセンブリ 発現量推定 ~ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
06.08. 版 スライド 8 までは自習 当日はスライド 9 から始める予定 スライド 3-86 は当日省略予定 講習会後に各自で復習してください 第 3 部 :NGS 解析 ( 中 ~ 上級 ) ~ トランスクリプトームアセンブリ 発現量推定 ~ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) kadota@iu.a.u-tokyo.ac.jp
我々のビッグデータ処理の新しい産業応用 広告やゲーム レコメンだけではない 個別化医療 ( ライフサイエンス ): 精神神経系疾患 ( うつ病 総合失調症 ) の網羅的ゲノム診断法の開発 全人類のゲノム解析と個別化医療実現を目標 ゲノム育種 ( グリーンサイエンス ): ブルーベリー オオムギ イネ
: 精神神経系疾患 ( うつ病 総合失調症 ) の網羅的ゲノム診断法の開発 全人類のゲノム解析と個別化医療実現を目標 ゲノム育種 ( グリーンサイエンス ): ブルーベリー オオムギ イネ") モンテカルロ法による分子進化の分岐図作成 のための最適化法 石井一夫 1 松田朋子 2 古崎利紀 1 後藤哲雄 2 1 東京農工大学 2 茨城大学 2013 9 9 2013 1 我々のビッグデータ処理の新しい産業応用 広告やゲーム レコメンだけではない 個別化医療 ( ライフサイエンス ): 精神神経系疾患 ( うつ病 総合失調症 ) の網羅的ゲノム診断法の開発 全人類のゲノム解析と個別化医療実現を目標
モンテカルロ法による分子進化の分岐図作成 のための最適化法 石井一夫 1 松田朋子 2 古崎利紀 1 後藤哲雄 2 1 東京農工大学 2 茨城大学 2013 9 9 2013 1 我々のビッグデータ処理の新しい産業応用 広告やゲーム レコメンだけではない 個別化医療 ( ライフサイエンス ): 精神神経系疾患 ( うつ病 総合失調症 ) の網羅的ゲノム診断法の開発 全人類のゲノム解析と個別化医療実現を目標
1 開発ツールのインストール 最初に JDK をインストールし 次に IDE をインストールする という手順になります 1. JDK のインストール JDK のダウンロードとインストール JDK は次の URL でオラクル社のウェブページからダウンロードします
 1 開発ツールのインストール 最初に JDK をインストールし 次に IDE をインストールする という手順になります 1. JDK のインストール JDK のダウンロードとインストール JDK は次の URL でオラクル社のウェブページからダウンロードします http://www.oracle.com/technetwork/java/javase/downloads/index.html なお
1 開発ツールのインストール 最初に JDK をインストールし 次に IDE をインストールする という手順になります 1. JDK のインストール JDK のダウンロードとインストール JDK は次の URL でオラクル社のウェブページからダウンロードします http://www.oracle.com/technetwork/java/javase/downloads/index.html なお
論文題目 腸管分化に関わるmiRNAの探索とその発現制御解析
 論文題目 腸管分化に関わる microrna の探索とその発現制御解析 氏名日野公洋 1. 序論 microrna(mirna) とは細胞内在性の 21 塩基程度の機能性 RNA のことであり 部分的相補的な塩基認識を介して標的 RNA の翻訳抑制や不安定化を引き起こすことが知られている mirna は細胞分化や増殖 ガン化やアポトーシスなどに関与していることが報告されており これら以外にも様々な細胞諸現象に関与していると考えられている
論文題目 腸管分化に関わる microrna の探索とその発現制御解析 氏名日野公洋 1. 序論 microrna(mirna) とは細胞内在性の 21 塩基程度の機能性 RNA のことであり 部分的相補的な塩基認識を介して標的 RNA の翻訳抑制や不安定化を引き起こすことが知られている mirna は細胞分化や増殖 ガン化やアポトーシスなどに関与していることが報告されており これら以外にも様々な細胞諸現象に関与していると考えられている
PowerPoint Presentation
 製品ソフトウェアのセットアップ手順 UNIX/Linux 編 1. セットアップファイルの選択開発環境 / 実行環境 / バージョン /Hotfix/ インストール先 OS 2. 対象セットアップファイルのダウンロード開発環境の場合は 2 つのファイルが対象 3. ソフトウェア要件の確認 4. ソフトウェアのインストール 5. ライセンスの認証 1 1. セットアップファイルの選択 選択項目選択肢該当チェック
製品ソフトウェアのセットアップ手順 UNIX/Linux 編 1. セットアップファイルの選択開発環境 / 実行環境 / バージョン /Hotfix/ インストール先 OS 2. 対象セットアップファイルのダウンロード開発環境の場合は 2 つのファイルが対象 3. ソフトウェア要件の確認 4. ソフトウェアのインストール 5. ライセンスの認証 1 1. セットアップファイルの選択 選択項目選択肢該当チェック
目次 第 1 章はじめに 本ソフトの概要... 2 第 2 章インストール編 ソフトの動作環境を確認しましょう ソフトをコンピュータにセットアップしましょう 動作を確認しましょう コンピュータからアンインストー
 JS 管理ファイル作成支援ソフト 設計用 Ver.4.0 インストールマニュアル 操作マニュアル 日本下水道事業団 目次 第 1 章はじめに... 1 1-1 本ソフトの概要... 2 第 2 章インストール編... 3 2-1 ソフトの動作環境を確認しましょう... 4 2-2 ソフトをコンピュータにセットアップしましょう... 5 2-3 動作を確認しましょう... 9 2-4 コンピュータからアンインストールする方法...
JS 管理ファイル作成支援ソフト 設計用 Ver.4.0 インストールマニュアル 操作マニュアル 日本下水道事業団 目次 第 1 章はじめに... 1 1-1 本ソフトの概要... 2 第 2 章インストール編... 3 2-1 ソフトの動作環境を確認しましょう... 4 2-2 ソフトをコンピュータにセットアップしましょう... 5 2-3 動作を確認しましょう... 9 2-4 コンピュータからアンインストールする方法...
プロジェクト概要 ー ヒト全遺伝子 データベース(H-InvDB)の概要と進展
の概要と進展") 個別要素技術 2 疾患との関連情報の抽出 予測のための 技術開発 平成 20 年 11 月 18 日産業技術総合研究所バイオメディシナル情報研究センター分子システム情報統合チーム 今西規 1 個別要素技術 2 課題一覧 1 大量文献からの自動知識抽出と文献からの既知疾患原因遺伝子情報の網羅的収集 2 疾患遺伝子情報整備と新規疾患遺伝子候補の予測 3 遺伝子多型情報整備 1 大量文献からの自動知識抽出と
個別要素技術 2 疾患との関連情報の抽出 予測のための 技術開発 平成 20 年 11 月 18 日産業技術総合研究所バイオメディシナル情報研究センター分子システム情報統合チーム 今西規 1 個別要素技術 2 課題一覧 1 大量文献からの自動知識抽出と文献からの既知疾患原因遺伝子情報の網羅的収集 2 疾患遺伝子情報整備と新規疾患遺伝子候補の予測 3 遺伝子多型情報整備 1 大量文献からの自動知識抽出と
Maeda140303
 2014 NGS NIBB - - - - FASTA / FASTQ - BED GFF/GTF WIG - SAM / BAM - SAMtools Web HTML (PC/), OS (Windows/Mac), IE/Chrome/Safari NGS Wet - - NGS - FASTA, FASTQ, csfastq, FASTA/qual, SRA, - BED, GFF/GTF,
2014 NGS NIBB - - - - FASTA / FASTQ - BED GFF/GTF WIG - SAM / BAM - SAMtools Web HTML (PC/), OS (Windows/Mac), IE/Chrome/Safari NGS Wet - - NGS - FASTA, FASTQ, csfastq, FASTA/qual, SRA, - BED, GFF/GTF,
PowerPoint Presentation
 Introduction to key concepts in Illumina sequencing data analysis イルミナシーケンスデータ解析入門その前に 癸生川絵里 (Eri Kibukawa) Bioinformatics Support Scientist 2012 Illumina, Inc. All rights reserved. Illumina, illuminadx,
Introduction to key concepts in Illumina sequencing data analysis イルミナシーケンスデータ解析入門その前に 癸生川絵里 (Eri Kibukawa) Bioinformatics Support Scientist 2012 Illumina, Inc. All rights reserved. Illumina, illuminadx,
<< 目次 >> 1 PDF コンバータのインストール ライセンスコードの入力 PDF にフォントを埋め込みたい場合の設定 PDF オートコンバータ EX で使用しない場合 PDF コンバータ単体で使用する場合の説明 PDF コンバータのアン
 PDF コンバータ V5.X インストール ガイド Page0 > 1 PDF コンバータのインストール... 2 2 ライセンスコードの入力... 6 3 PDF にフォントを埋め込みたい場合の設定... 9 4 PDF オートコンバータ EX で使用しない場合 PDF コンバータ単体で使用する場合の説明... 10 5 PDF コンバータのアンインストール... 16 6 お問合せ...
PDF コンバータ V5.X インストール ガイド Page0 > 1 PDF コンバータのインストール... 2 2 ライセンスコードの入力... 6 3 PDF にフォントを埋め込みたい場合の設定... 9 4 PDF オートコンバータ EX で使用しない場合 PDF コンバータ単体で使用する場合の説明... 10 5 PDF コンバータのアンインストール... 16 6 お問合せ...
Acrobat Reader DCのインストール・操作方法―Windows 10/8.1/7
 PDF 閲覧ソフトの定番ソフトです ダウンロード インストールと基本的な操作方法を図解します Windows 10, 8.1 及び 7 での操作の図解です Windows 10 でインストールし操作する方法図解 Windows 8.1 でのインストールの方法図解 Windows 7 でのインストールの方法図解 古いバージョンを使いたい時は Acrobat Reader XI, X 性能が良くてタブ式の
PDF 閲覧ソフトの定番ソフトです ダウンロード インストールと基本的な操作方法を図解します Windows 10, 8.1 及び 7 での操作の図解です Windows 10 でインストールし操作する方法図解 Windows 8.1 でのインストールの方法図解 Windows 7 でのインストールの方法図解 古いバージョンを使いたい時は Acrobat Reader XI, X 性能が良くてタブ式の
24th Embarcadero Developer Camp
 17 Th Developer Camp B4 Delphi/C++Builder テクニカルワークショップ Delphi / C++Builder 旧バージョンアプリケーションの移行 エンバカデロ テクノロジーズサポートチーム with 高橋智宏 1 17 Th Developer Camp Delphi Q1 2 midas.dll Q. 別々のバージョンで作成したデータベースアプリケーションがあります
17 Th Developer Camp B4 Delphi/C++Builder テクニカルワークショップ Delphi / C++Builder 旧バージョンアプリケーションの移行 エンバカデロ テクノロジーズサポートチーム with 高橋智宏 1 17 Th Developer Camp Delphi Q1 2 midas.dll Q. 別々のバージョンで作成したデータベースアプリケーションがあります
免疫形式文法
 遺伝子発現解析入門 中岡慎治 目次 はじめに 遺伝子発現 ( トランスクリプトーム ) 解析とはマイクロアレイ (MA) の原理と応用途次世代シーケンサー (NGS) の原理と応用途 [ 補足 ] 次世代シーケンサーの活用事例 [metagenome/chip-seq] etc 遺伝子発現解析の統計手法 正規化の必要性と手法 [MA/NGS] 発現変動解析 (Differential Expressed
遺伝子発現解析入門 中岡慎治 目次 はじめに 遺伝子発現 ( トランスクリプトーム ) 解析とはマイクロアレイ (MA) の原理と応用途次世代シーケンサー (NGS) の原理と応用途 [ 補足 ] 次世代シーケンサーの活用事例 [metagenome/chip-seq] etc 遺伝子発現解析の統計手法 正規化の必要性と手法 [MA/NGS] 発現変動解析 (Differential Expressed
Microsoft PowerPoint - GLMMexample_ver pptx
 Linear Mixed Model ( 以下 混合モデル ) の短い解説 この解説のPDFは http://www.lowtem.hokudai.ac.jp/plantecol/akihiro/sumida-index.html の お勉強 のページにあります. ver 20121121 と との間に次のような関係が見つかったとしよう 全体的な傾向に対する回帰直線を点線で示した ところが これらのデータは実は異なる
Linear Mixed Model ( 以下 混合モデル ) の短い解説 この解説のPDFは http://www.lowtem.hokudai.ac.jp/plantecol/akihiro/sumida-index.html の お勉強 のページにあります. ver 20121121 と との間に次のような関係が見つかったとしよう 全体的な傾向に対する回帰直線を点線で示した ところが これらのデータは実は異なる
Shareresearchオンラインマニュアル
 Chrome の初期設定 以下の手順で設定してください 1. ポップアップブロックの設定 2. 推奨する文字サイズの設定 3. 規定のブラウザに設定 4. ダウンロードファイルの保存先の設定 5.PDFレイアウトの印刷設定 6. ランキングやハイライトの印刷設定 7. 注意事項 なお 本マニュアルの内容は バージョン 61.0.3163.79 の Chrome を基に説明しています Chrome の設定手順や画面については
Chrome の初期設定 以下の手順で設定してください 1. ポップアップブロックの設定 2. 推奨する文字サイズの設定 3. 規定のブラウザに設定 4. ダウンロードファイルの保存先の設定 5.PDFレイアウトの印刷設定 6. ランキングやハイライトの印刷設定 7. 注意事項 なお 本マニュアルの内容は バージョン 61.0.3163.79 の Chrome を基に説明しています Chrome の設定手順や画面については
Ontrack EasyRecovery 11 基本的な使い方
 1.Ontrack EasyRecovery 11 の基本的な使い方 1Ontrack EasyRecovery 11 を起動し 画面右下の 次へ をクリックします ここの画面 操作は Ontrack EasyRecovery 11 Home for Windows を使用した例となります 2 下記の メディアの種類を選択 の画面になりますので 復旧対象のディスクを選択し 次へ をクリックしてください
1.Ontrack EasyRecovery 11 の基本的な使い方 1Ontrack EasyRecovery 11 を起動し 画面右下の 次へ をクリックします ここの画面 操作は Ontrack EasyRecovery 11 Home for Windows を使用した例となります 2 下記の メディアの種類を選択 の画面になりますので 復旧対象のディスクを選択し 次へ をクリックしてください
Windows10の標準機能だけでデータを完全バックアップする方法 | 【ぱそちき】パソコン初心者に教えたい仕事に役立つPC知識
 ぱそちき パソコン初心者に教えたい仕事に役立つ PC 知識 Windows10 の標準機能だけでデータを完全バックアッ プする方法 パソコンが急に動かなくなったり 壊れてしまうとパソコンに保存していたテキストや写真などの データも無くなってしまいます このように思いがけない事故からデータを守るには バックアップを取っておくしかありません Windows10のパソコンを使っているなら データをバックアップするのに特別なソフトは必要ありません
ぱそちき パソコン初心者に教えたい仕事に役立つ PC 知識 Windows10 の標準機能だけでデータを完全バックアッ プする方法 パソコンが急に動かなくなったり 壊れてしまうとパソコンに保存していたテキストや写真などの データも無くなってしまいます このように思いがけない事故からデータを守るには バックアップを取っておくしかありません Windows10のパソコンを使っているなら データをバックアップするのに特別なソフトは必要ありません
kiso2-03.key
 座席指定はありません Linux を起動して下さい 第3回 計算機基礎実習II 2018 のウェブページか ら 以下の課題に自力で取り組んで下さい 計算機基礎実習II 第2回の復習課題(rev02) 第3回の基本課題(base03) 第2回課題の回答例 ex02-2.c include int main { int l int v, s; /* 一辺の長さ */ /* 体積 v
座席指定はありません Linux を起動して下さい 第3回 計算機基礎実習II 2018 のウェブページか ら 以下の課題に自力で取り組んで下さい 計算機基礎実習II 第2回の復習課題(rev02) 第3回の基本課題(base03) 第2回課題の回答例 ex02-2.c include int main { int l int v, s; /* 一辺の長さ */ /* 体積 v
バイオインフォマティクスⅠ
 バイオインフォマティクス ( 第 5 回 ) 慶應義塾大学生命情報学科 榊原康文 多重アライメントの解 0 2 3 4 5 6 7 j Q T S Y T R Y Q T - Y T R K 0 0-9 -20-44 -52-63 -72-90 Q -6 2 0-6 -4-25 -34-52 2 S -32 5 30 4 6-5 -4-32 3 Y -48-4 2 38 27 8 0 4 P -64-27
バイオインフォマティクス ( 第 5 回 ) 慶應義塾大学生命情報学科 榊原康文 多重アライメントの解 0 2 3 4 5 6 7 j Q T S Y T R Y Q T - Y T R K 0 0-9 -20-44 -52-63 -72-90 Q -6 2 0-6 -4-25 -34-52 2 S -32 5 30 4 6-5 -4-32 3 Y -48-4 2 38 27 8 0 4 P -64-27
Chromeleon 6 for Chromeleon 6.8 SR15 Build: --- 新しいシーケンスの作成に使用できるワークリストファイル (.wle) Doc. Nr: CM6_68150_0020 Doc. Ver.: Doc. Type: Guide
 Doc. Nr: CM6_68150_0020 Doc. Ver.: Doc. Type: Guide") for.8 SR15 Build: --- LIMS ワークリストの書式 はじめに Chromeleon における LIMS データ ( シーケンス ) 読取りフォーマットをワークリスト (WLE ファイル ) といいます ワークリストファイル形式で LIMS から情報を出力して頂ければ Chromeleon でインポートできます そのため LIMS からワークリスト形式で出力できるように LIMS
for.8 SR15 Build: --- LIMS ワークリストの書式 はじめに Chromeleon における LIMS データ ( シーケンス ) 読取りフォーマットをワークリスト (WLE ファイル ) といいます ワークリストファイル形式で LIMS から情報を出力して頂ければ Chromeleon でインポートできます そのため LIMS からワークリスト形式で出力できるように LIMS
Maser RNA-seq Genome Resequencing De novo Genome Sequencing Metagenome ChIP-seq CAGE BS-seq
 NGS Maser 2013/10/17 Maser RNA-seq Genome Resequencing De novo Genome Sequencing Metagenome ChIP-seq CAGE BS-seq Maser RNA-seq Genome Resequencing De novo Genome Sequencing Metagenome ChIP-seq CAGE BS-seq
NGS Maser 2013/10/17 Maser RNA-seq Genome Resequencing De novo Genome Sequencing Metagenome ChIP-seq CAGE BS-seq Maser RNA-seq Genome Resequencing De novo Genome Sequencing Metagenome ChIP-seq CAGE BS-seq
Maple 12 Windows版シングルユーザ/ネットワークライセンス
 Maple Network Tools インストール 設定手順書 更新日 2017/07/27 はじめに この手順書は Windows 32bit Windows 64bit Mac OS Linux に対応しております 詳しい動作環境については こちらを参照願います http://www.cybernet.co.jp/maple/product/system/maple.html この手順書の説明画面は
Maple Network Tools インストール 設定手順書 更新日 2017/07/27 はじめに この手順書は Windows 32bit Windows 64bit Mac OS Linux に対応しております 詳しい動作環境については こちらを参照願います http://www.cybernet.co.jp/maple/product/system/maple.html この手順書の説明画面は
SOC Report
 Apache-Tomcat と冗長な UTF-8 表現 (CVE-2008-2938 検証レポート ) N T T コミュニケーションズ株式会社 IT マネジメントサービス事業部セキュリティオペレーションセンタ 2009 年 5 月 26 日 Ver. 1.1 1. 調査概要... 3 2. UTF-8 とは... 3 3. CVE-2008-2938... 4 3.1..( ピリオド ) について...
Apache-Tomcat と冗長な UTF-8 表現 (CVE-2008-2938 検証レポート ) N T T コミュニケーションズ株式会社 IT マネジメントサービス事業部セキュリティオペレーションセンタ 2009 年 5 月 26 日 Ver. 1.1 1. 調査概要... 3 2. UTF-8 とは... 3 3. CVE-2008-2938... 4 3.1..( ピリオド ) について...
PDFオートコンバータEX
 PDF コンバータ V4.X インストール ガイド Page0 > 1 PDF コンバータ 32BIT 版のインストール... 2 2 PDF コンバータ 64BIT 版のインストール... 7 3 PDF にフォントを埋め込みたい場合の設定... 13 4 PDF オートコンバータ EX で使用しない場合 PDF コンバータ単体で使用する場合の説明... 14 5 PDF コンバータのアンインストール...
PDF コンバータ V4.X インストール ガイド Page0 > 1 PDF コンバータ 32BIT 版のインストール... 2 2 PDF コンバータ 64BIT 版のインストール... 7 3 PDF にフォントを埋め込みたい場合の設定... 13 4 PDF オートコンバータ EX で使用しない場合 PDF コンバータ単体で使用する場合の説明... 14 5 PDF コンバータのアンインストール...
PowerPoint プレゼンテーション
 JAN コード登録マニュアル 項目説明 CSV で商品データを upload するに当たり 間違えやすいカラムについてまとめました 項目 説明 備考 コントロールカラム CSV 上で当該商品情報をどうするのか ( 更新 削除等 ) 指示するコード "u": 更新 "d": 削除等 商品管理番号 出来上がった商品ページURLの一部であり 入力がない場合は自動採番される web 上で商品を特定するキーコード
JAN コード登録マニュアル 項目説明 CSV で商品データを upload するに当たり 間違えやすいカラムについてまとめました 項目 説明 備考 コントロールカラム CSV 上で当該商品情報をどうするのか ( 更新 削除等 ) 指示するコード "u": 更新 "d": 削除等 商品管理番号 出来上がった商品ページURLの一部であり 入力がない場合は自動採番される web 上で商品を特定するキーコード
Slide 1
 NGS をはじめよう!RNA-Seq 入門 ( キットの選び方 実験デザイン ) April 18, 2014 米田瑞穂イルミナ株式会社テクニカルアプリケーションサイエンティスト 2012 Illumina, Inc. All rights reserved. Illumina, illuminadx, BaseSpace, BeadArray, BeadXpress, cbot, CSPro, DASL,
NGS をはじめよう!RNA-Seq 入門 ( キットの選び方 実験デザイン ) April 18, 2014 米田瑞穂イルミナ株式会社テクニカルアプリケーションサイエンティスト 2012 Illumina, Inc. All rights reserved. Illumina, illuminadx, BaseSpace, BeadArray, BeadXpress, cbot, CSPro, DASL,
WebSAM System Navigator JNS isadmin SNMP Trap 連携設定手順書 NEC 2012 年 12 月
 WebSAM System Navigator JNS isadmin SNMP Trap 連携設定手順書 NEC 202 年 2 月 目次. はじめに 2. ナレッジの格納 3.WebSAMSystemNavigator の初期設定 4. トポロジビューの設定 5. ビジネスビューの設定 6. メッセージの表示 Page 2 NEC Corporation 202 . はじめに 本書は JNS 株式会社の
WebSAM System Navigator JNS isadmin SNMP Trap 連携設定手順書 NEC 202 年 2 月 目次. はじめに 2. ナレッジの格納 3.WebSAMSystemNavigator の初期設定 4. トポロジビューの設定 5. ビジネスビューの設定 6. メッセージの表示 Page 2 NEC Corporation 202 . はじめに 本書は JNS 株式会社の