特論I
|
|
|
- そうすけ ほうねん
- 7 years ago
- Views:
Transcription


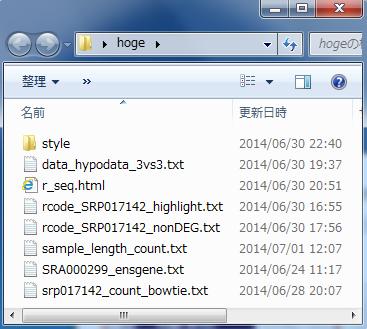
1 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 4 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 [email protected] 1
2 前回の課題と正答 アダプター配列除去前後の small RNA-seq データをカイコゲノムにマップし マップ率 ( マップされたリード数 ) を比較する 1. マッピング前の総リード数を示せ アダプター配列除去前のSRR fastq.gz: 11,928,428 リード アダプター配列除去後のhoge4.fastq.gz: 11,928,428 リード 2. マッピング後の マップされたリード数 を示せ アダプター配列除去前のSRR fastq.gz: アダプター配列除去後のhoge4.fastq.gz: 3. 結果の考察 2,257 リード 1,308,126 リード マッピング後の総リード数ではなく マップされたリード数が正解ですね 失礼しました 2
3 Contents( 第 4 回 ) 新規転写物同定 ( ゲノム情報を利用 ) 基本的な考え方 Tophat-Cufflinks パイプライン 可視化 ( ゲノムブラウザや Viewer) 発現量推定 ( 遺伝子レベルと転写物レベル ) RPKM の基本的な考え方 計算時間短縮戦略 ( トランスクリプトーム情報のみを利用 ) カウントデータを用いたサンプル間比較解析 イントロ ( カウントデータ取得まで ) サンプル間クラスタリング 発現変動遺伝子検出 分布やモデル 課題 3
4 トランスクリプトーム解析の目的は様々 トランスクリプトーム配列取得 ゲノム配列既知の場合 :Cufflinksなどを用いて遺伝子構造推定( アノテーション ) ゲノム配列未知の場合 :Trinityなどのトランスクリプトーム用アセンブラを実行 遺伝子または転写物 (isoform) ごとの発現量の正確な推定 RSEMなどを利用して発現量情報を得る ある特定のサンプル内での遺伝子間の発現量の大小関係を知りたい 配列長やGC biasなどの各種補正がポイント 比較するサンプル間で発現変動している遺伝子または転写物の同定 TCCパッケージなどを利用して発現変動遺伝子 (DEG) を得る ライブラリサイズ ( 総リード数 ) や発現している遺伝子の組成の補正がポイント (GO 解析など )DEG 結果を用いる多くの下流解析結果に影響を及ぼす 4
 はマップされないので")
5 マッピングの基本的なイメージ 基本的なマッピングプログラム (bowtie など ) を用いた場合 教科書 p81-89 リファレンス配列 : ゲノム count T1 サンプルの RNA-Seq データ mapping 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 リファレンス配列 : トランスクリプトーム count 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 ゲノム配列へのマッピングの場合 複数のエクソンにまたがるリード (spliced reads) はマップされないので 5
6 RNA-MATE (Cloonan et al., Bioinformatics, 25: , 2009) 対策 ( リード長が 75bp 程度以上の現在 ) 再帰的にマッピングする戦略 (recursive mapping strategy) 通常のマッピングプログラムでマップされなかったものに対して リードを短くしてマップされるかどうかを繰り返すというイメージ >75bp 程度のマップされなかったリードの集団 mapping 遺伝子 1 マップされない 遺伝子 1 マップされない 遺伝子 1 マップされた splice-aware aligner (spliced aligner) を用いることで新規転写物の同定も可能 理由は既知遺伝子構造情報を参照しなくてもどうにかなるから 6
7 Splice-aware aligner の様々な戦略 Garber et al., Nat. Methods, 8: , 2011 の Fig. 1 exon-first 系は高速だがアルゴリズム的に processed pseudogene 存在下で正確な構造推定が困難になる 7

8 Basic aligner (unspliced aligner) Windows でマッピング可能な R パッケージ 内部的に basic aligner の bowtie と splice-aware aligner の SpliceMap を利用可能 比較的よく使われているもの 8
")
9 Splice-aware aligner (spliced aligner) Windows でマッピング可能な R パッケージ 内部的に basic aligner の bowtie と splice-aware aligner の SpliceMap を利用可能 比較的よく使われているもの Tophat は内部的に Bowtie を利用 ( 今は Bowtie 2 かも ) 9
10 Reference-based strategy Splice-aware aligner 出力結果をもとに遺伝子構造推定 Scripture (Guttman et al., Nat. Biotechnol., 28: , 2010) Cufflinks (Trapnell et al., Nat. Biotechnol., 28: , 2010) STM (Surget-Groba and Montoya-Burgos, Genome Res., 20: , 2010) ALEXA-seq (Griffith et al., Nat. Methods, 7: , 2010) ARTADE2 (Kawaguchi et al., Bioinformatics, 28: , 2012) この transcriptome reconstruction 作業は結構大変 理由 1: 広いダイナミックレンジ ( 低発現のものとノイズとの区別 ) 理由 2:off-targetの存在 (mature mrna 以外のprecursor RNAなど ) 理由 3: 一つの遺伝子から複数のisoforms( どのisoform 由来のリードか?!) exon a gene (or a locus) isoform1 isoform2 isoform3 10
11 Martin and Wang, Nature Reviews Genet., 12: , 2011 の Fig. 2 遺伝子構造推定のイメージ 11



12 Bowtie や Tophat が多く引用されるのは Cufflinks など他のソフトウェア上でもよく実装されているためであろう 12
13 Bowtie-Tophat-Cufflinks パイプライン basic aligner splice-aware aligner Trapnell et al., Nat. Protoc., 7: , 2012 Fig. 1 Fig. 2 RNA-seq データとリファレンス配列情報を入力として 遺伝子構造推定から発現量 発現変動解析 描画までの一連の解析を提供 13
14 Bowtie-Tophat-Cufflinks パイプライン Fig. 2 Trapnell et al., Nat. Protoc., 7: , 2012 Fig. 3 RNA-seq データとリファレンス配列情報を入力として 遺伝子構造推定から発現量 発現変動解析 描画までの一連の解析を提供 14


15 NGS データ解析手段 自前で大容量メモリ計算サーバ (Linux) を購入し 必要なソフトのインストールからスタート 難易度は高いが思い通りの解析が可能 Linux サーバをもつバイオインフォ系の人にお願いする 気軽に頼める知り合いがいればいいが その人次第 DDBJ Read Annotation Pipeline を利用 一番お手軽な選択肢であり 有名どころはカバーされている Cufflinks もできます 15
 可視化ツールは全く使いません")
16 可視化 ( ゲノムブラウザや Viewer) 私は ( 数値解析系なので ) 可視化ツールは全く使いません 比較的よく使われているもの 16
 私は (")

17 可視化 ( ゲノムブラウザや Viewer) 私は ( 数値解析系なので ) 可視化ツールは全く使いません 比較的よく使われているもの 17
18 Contents( 第 4 回 ) 新規転写物同定 ( ゲノム情報を利用 ) 基本的な考え方 Tophat-Cufflinks パイプライン 可視化 ( ゲノムブラウザや Viewer) 発現量推定 ( 遺伝子レベルと転写物レベル ) RPKM の基本的な考え方 計算時間短縮戦略 ( トランスクリプトーム情報のみを利用 ) カウントデータを用いたサンプル間比較解析 イントロ ( カウントデータ取得まで ) サンプル間クラスタリング 発現変動遺伝子検出 分布やモデル 課題 18
19 マップされたリード数 = 発現量ではないが 基本的なマッピングプログラム (bowtie など ) を用いた場合 リファレンス配列 : ゲノム count G G1 サンプルの RNA-Seq データ mapping 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 リファレンス配列 : トランスクリプトーム count G 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 マップされたリード数のカウント情報は 発現量推定の基本情報です 19
20 研究目的別留意点 : 遺伝子間比較 教科書 p 発現量補正の基本形 : RPK (Reads per kilobase) RPM (Reads per million) RPKM (Reads per kilobase per million) 定数カウント数 配列長 総リード数 同一サンプル内での異なる遺伝子間の発現レベル比較の場合 配列長由来 bias: 長いほど沢山 sequence される RPKM や FPKM などの配列長を考慮して正規化されたデータで解析 GC 含量由来 bias: カウント数の分布が GC 含量依存的である Risso et al., BMC Bioinformatics, 12: 480, 2011 Benjamini and Speed, Nucleic Acids Res., 40: e72, 2012 Filloux et al., BMC Bioinformatics, 15: 188, 2014 総リード数 ( ライブラリサイズ or sequence depth) 補正は不必要理由 : 遺伝子間の発現レベルの大小関係は定数倍しても不変 20
21 研究目的別留意点 : サンプル間比較 発現量補正の基本形 : RPK (Reads per kilobase) RPM (Reads per million) RPKM (Reads per kilobase per million) 異なるサンプル間での同一遺伝子間の発現レベル比較の場合 総リード数の違い : 総リード数が x 倍違うと全体的に x 倍変動 RPM 正規化で全体を揃えることは基本 定数カウント数 配列長 総リード数 組成の違い : サンプル特異的高発現遺伝子の存在で比較困難に TMM 正規化法 (Robinson and Oshlack, Genome Biol., 11: R25, 2010) TbT 正規化法 (Kadota et al., Algorithms Mol. Biol., 7: 5, 2012) DEGES に基づく正規化法 (Sun et al., BMC Bioinformatics, 14: 219, 2013) 配列長や GC bias 補正は少なくとも理論上は不必要理由 : 同一遺伝子に対して掛かる係数はサンプル間で同じ 教科書 p
22 配列長の補正 教科書 p Mortazavi et al., Nat. Methods, 5: , 2008 配列長が長い遺伝子ほど沢山 sequence される それらの遺伝子上にマップされる生のリード数が増加傾向配列長が長い遺伝子ほど発現レベルが高い傾向になる 発現レベルが同じで長さの異なる二つの mrnas AAAAAAA AAAAAAA 断片化して sequence マップされたリード数をカウント AAAAAAA AAAAAAA 1 つのサンプル内で異なる遺伝子間の発現レベルの大小関係を配列長を考慮せずに比較することはできない 22
23 教科書 p 配列長を考慮した発現量推定のイメージ gene1: 3 exons (middle length), 14 reads mapped (low coverage) gene2: 3 exons (middle length), 56 reads mapped (high coverage) gene3: 2 exons (short length), 12 reads mapped (middle coverage) gene4: 2 exons (long length), 31 reads mapped (middle coverage) マップされたリード分布生リードカウント結果補正度の発現量 Garber et al., Nat. Methods, 8: , 2011 の Fig. 3a 長さが同じならリード数の多い方が発現量高い (gene 1 対 2) 長いほどマップされるリード数が多くなる効果を補正する必要がある (gene 3 対 4) 1 つのサンプル内で転写物または遺伝子間の発現レベルの大小を比較したい場合には配列長を考慮すべきである 23

 2")
24 配列長とカウント数の関係を眺める 入力ファイル読み込み時にrow.names=1 としているので dataオブジェクトの1 列目がwidth 列 ( 配列長情報 ) 2 列目がKidney 列 ( 腎臓サンプルのカウント情報 ) となる 24

25 配列長とカウント数の関係を眺める 数値のダイナミックレンジが広いので x 軸 y 軸ともに log10 変換してプロットしている 0 カウントのものは log をとれない関係上 プロットできないという警告が出ています 確かに水平ではなく全体的に右斜め上になっている傾向が見られます 25

26 配列長とカウント数の関係を眺める ただの検証ですが ゼロカウントデータが相当数存在することが分かります 26



27 配列長順にソートし カウント数を 20 分割したものを boxplot で示したもの 様々な表現手段があります 27
 に相当 Reads Per Kilobase (RPK) Counts Per Kilobase")
28 配列長の補正 前提条件 : 配列長が既知 補正の基本戦略 : 配列長で割る 1 / 配列長 を掛ける場合 教科書 p 塩基あたりの平均のリード数 の計算に相当 1000 / 配列長 を掛ける場合 Mortazavi et al., Nat. Methods, 5: , 2008 AAAAAAA AAAAAAA その遺伝子の配列長が 1000bp だったときのリード数 (or カウント数 ) に相当 Reads Per Kilobase (RPK) Counts Per Kilobase (CPK) 28
 になるような正規化係数を掛ける例 :sample1 の正規化係数 = 100 /")
29 マイクロアレイデータの正規化 参考 各サンプルから測定されたシグナル強度の和は一定 アレイ上の遺伝子数が少ない場合は非現実的だが 数千 ~ 数万種類の遺伝子が搭載されているので妥当という思想 グローバル正規化 背景 : サンプルごとにシグナル強度の総和は異なる対策 : 総和が任意の値 ( 例では 100) になるような正規化係数を掛ける例 :sample1 の正規化係数 = 100 /
 正規化後の総リード数が 100 万 (one million) になるように補正例 :T1 の正規化係数 = 1000000 / 67")
30 RNA-Seq データの正規化の一部 発現している RNA 量の総和はサンプル間で一定 教科書 p 参考 遺伝子 1 遺伝子 2 遺伝子 3 遺伝子 4 RPM 正規化 Reads Per Million mapped reads(rpm) 正規化後の総リード数が 100 万 (one million) になるように補正例 :T1 の正規化係数 = / 67 30
 配列長が 1,000 bp かつ総リード数が 100 万だったときのカウント数 RPKM 1,000 1,000,000")

31 RPKM 教科書 p Reads per kilobase (of exon) per million (mapped reads) 配列長が 1,000 bp かつ総リード数が 100 万だったときのカウント数 RPKM 1,000 1,000,000 カウント数 配列長総リード数 1,000,000,000 カウント数 配列長 総リード数 sample_length_count.txt hoge1.txt 総リード数 = 教科書の説明もみながら RPK, RPM, RPKM の例題を実行しておきましょう 31
32 少ない カウント数 多い EDASeq(Risso et al., BMC Bioinformatics, 12: 480, 2011) の Fig.1 GC bias 補正の必要性も提唱されている 参考 GC 含量が多い遺伝子や少ない遺伝子上にマップされたリードカウント数は GC 含量が中程度の遺伝子に比べて少ない傾向にある 少ない 多い 32
33 EDASeq(Risso et al., BMC Bioinformatics, 12: 480, 2011) の Fig.1 参考 GC bias 補正の必要性も提唱されている Quantile 正規化 パッケージ中のサンプルファイルを解析してみると 確かに GC bias が緩和されていることがわかる 33
34 Contents( 第 4 回 ) 新規転写物同定 ( ゲノム情報を利用 ) 基本的な考え方 Tophat-Cufflinks パイプライン 可視化 ( ゲノムブラウザや Viewer) 発現量推定 ( 遺伝子レベルと転写物レベル ) RPKM の基本的な考え方 計算時間短縮戦略 ( トランスクリプトーム情報のみを利用 ) カウントデータを用いたサンプル間比較解析 イントロ ( カウントデータ取得まで ) サンプル間クラスタリング 発現変動遺伝子検出 分布やモデル 課題 34


35 高速に発現量推定するための様々な戦略 ゲノム配列を利用するが アノテーション情報も同時に読み込んで発現量を得たい特定の領域のみにマッピングして高速化 :Cufflinks トランスクリプトーム転写物配列にマッピング :NEUMA, IsoEM, RSEM k-mer を用いた alignment-free な方法 :Sailfish, RNA-Skim トランスクリプトーム配列へのマッピングは bowtie のような basic aligner で必要十分 しかしマッピングが律速であるため alignmentfree な方法が注目されはじめている 35

36 転写物配列にマップして高速に発現量推定 Bowtie + express で高精度な結果を追求 (~days) RNA-Skim で超高速にそこそこの精度で定量化 (~min) 1 day = 60*60*24 = 86,400 seconds Zhang and Wang, Bioinformatics, 30: i283-i292, 2014 の Table 3 36
37 Contents( 第 4 回 ) 新規転写物同定 ( ゲノム情報を利用 ) 基本的な考え方 Tophat-Cufflinks パイプライン 可視化 ( ゲノムブラウザや Viewer) 発現量推定 ( 遺伝子レベルと転写物レベル ) RPKM の基本的な考え方 計算時間短縮戦略 ( トランスクリプトーム情報のみを利用 ) カウントデータを用いたサンプル間比較解析 イントロ ( カウントデータ取得まで ) サンプル間クラスタリング 発現変動遺伝子検出 分布やモデル 課題 37
38 カウントデータを用いたサンプル間比較解析 複製あり 2 群間比較用ヒト RNA-seq データ (3 Ras 対 3 Proliferative) カウントデータ 59,857 genes データ解析の基本イメージ 発現変動遺伝子 (DEG) 同定 サンプル間クラスタリング G1 群 G2 群 38
39 イントロ ( カウントデータ取得まで ) Step1: SRAdb を用いた gzip 圧縮 FASTQ 形式ファイルのダウンロード Neyret-Kahn et al., Genome Res., 23: , 2013 複製あり 2 群間比較用ヒト RNA-seq データ (3 Ras vs. 3 Proliferative) FileName SampleName SRR fastq.gz Pro_rep1 SRR fastq.gz Pro_rep2 SRR fastq.gz Pro_rep3 SRR fastq.gz Ras_rep1 SRR fastq.gz Ras_rep2 SRR fastq.gz Ras_rep3 G1 群 G2 群 1 つの論文中で ChIP-seq もやっており RNA-seq データのみダウンロードする際にちょっと困る例を紹介 39

 もちろん主観ですが ENA (ArrayExpress) よりも GEO")
40 イントロ ( カウントデータ取得まで ) Step1: SRAdb を用いた gzip 圧縮 FASTQ 形式ファイルのダウンロード Neyret-Kahn et al., Genome Res., 23: , 2013 複製あり 2 群間比較用ヒト RNA-seq データ (3 Ras vs. 3 Proliferative) もちろん主観ですが ENA (ArrayExpress) よりも GEO のほうがわかりやすいという特殊事例です 40


41 実データ解析例 :SRP ChIP-seq と RNA-seq 両方を 1 つの論文中でやっている場合には 論文と 1 対 1 対応の GSE42213 以外に さらに下の階層の GSE ID が付与されている GSE42211:ChIP-seq データ GSE42212:RNA-seq データ 41

42 ENA (ArrayExpress) の場合は Step1: SRAdb を用いた gzip 圧縮 FASTQ 形式ファイルのダウンロード Neyret-Kahn et al., Genome Res., 23: , 2013 複製あり2 群間比較用ヒトRNA-seqデータ (3 Ras vs. 3 Proliferative) ArrayExpressで眺めると サブシリーズのGSE ID (GSE42211 とGSE42212) が見当たらない 42

 をサブシリーズに分割せずに一覧可能にしたのはいいと思うが なぜ 26 サンプルが 34")
43 ENA (ArrayExpress) の場合は Neyret-Kahn et al., Genome Res., 23: , 2013 複製あり 2 群間比較用ヒト RNA-seq データ (3 Ras vs. 3 Proliferative) ChIP-seq データと RNA-seq データ (GSE42211 と GSE42212) をサブシリーズに分割せずに一覧可能にしたのはいいと思うが なぜ 26 サンプルが 34 になっているのか不明 43

44 イントロ ( カウントデータ取得まで ) Step1: 計 6 ファイル (2 群間比較用 ) FileName SampleName SRR fastq.gz Pro_rep1 SRR fastq.gz Pro_rep2 SRR fastq.gz Pro_rep3 SRR fastq.gz Ras_rep1 SRR fastq.gz Ras_rep2 SRR fastq.gz Ras_rep3 G1 群 G2 群 44

45 イントロ ( カウントデータ取得まで ) Step2: QuasR を用いたヒトゲノムへのマッピング リファレンス配列として BSgenome.Hsapiens.UCSC.hg19 という R パッケージを利用 約 18 生物種のゲノム配列が R パッケージとして利用可能シロイヌナズナ :BSgenome.Athaliana.TAIR.TAIR9 ショウジョウバエ :BSgenome.Dmelanogaster.UCSC.dm3 45


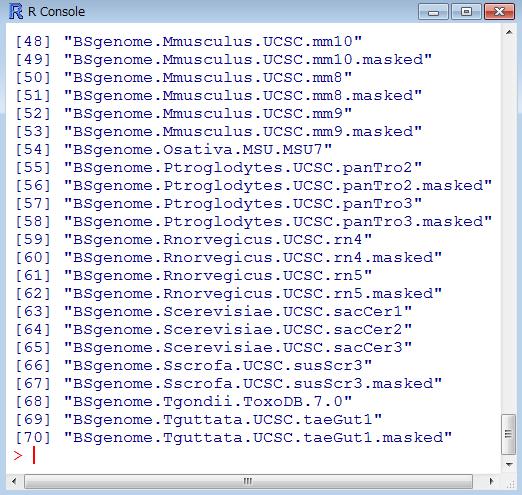
46 ゲノム配列の R パッケージがあります R および Bioconductor の最新版をインストールしたヒトが mm10 などゲノム配列の最新版も利用できます 定期的なバージョンアップの意義 46
47 Contents( 第 4 回 ) 新規転写物同定 ( ゲノム情報を利用 ) 基本的な考え方 Tophat-Cufflinks パイプライン 可視化 ( ゲノムブラウザや Viewer) 発現量推定 ( 遺伝子レベルと転写物レベル ) RPKM の基本的な考え方 計算時間短縮戦略 ( トランスクリプトーム情報のみを利用 ) カウントデータを用いたサンプル間比較解析 イントロ ( カウントデータ取得まで ) サンプル間クラスタリング 発現変動遺伝子検出 分布やモデル 課題 47
 は存在すると判断")
48 教科書 p サンプル間クラスタリング Pro 群と Ras 群に明瞭に分かれているので発現変動遺伝子 (DEG) は存在すると判断 フィルタリングの思想は教科書を参照 48
 と判定されたものが多数存在することがわかる 49")
49 教科書 p 発現変動遺伝子検出 発現変動遺伝子 (DEG) と判定されたものが多数存在することがわかる 49
50 教科書 p 発現変動遺伝子検出 5% 偽物を含むのを許容すると DEG 数は 5,669 個 20% の偽物混入を許容すると 8,110 DEGs FDR 閾値が 30% の場合は 9,151 個 このデータセット中に存在する本物の DEG は 9, = 6,405.7 個程度だと判断できる 論文に記載すべきデータ解析環境の情報 50
 横軸が平均(Average) G1 群 < G2 群 G2 群で高発現 G1 群 = G2 群 G1 群 > G2 群 G1 群で高発現 1 2 3 4 5 A =")
51 M = log 2 G2 - log 2 G M-A plot 教科書 p Dudoit et al., Stat. Sinica, 12: , 群間比較用 横軸が全体的な発現レベル 縦軸がlog 比からなるプロット 名前の由来は おそらく対数の世界での縦軸が引き算 (Minus) 横軸が平均(Average) G1 群 < G2 群 G2 群で高発現 G1 群 = G2 群 G1 群 > G2 群 G1 群で高発現 A = (log 2 G2 + log 2 G1)/2 低発現 全体的に 高発現 DEG が存在しないデータの M-A plot を眺めることで 縦軸の閾値のみに相当する倍率変化を用いた DEG 同定の危険性が分かります 51

52 発現変動遺伝子検出結果 TCC を用いた DEG 同定結果ファイル p-value とその順位 G2 群で高発現 M-A plot の A 値と M 値 q-value FDR 閾値判定結果 q-value < 0.05 を満たす DEG が 1 non-deg が 0 G1 群で高発現 基本的には これらが解析結果です 1 位は Ras 群 (G2 群 ) で高発現の DEG 52
53 発現変動遺伝子検出結果 TCC を用いた DEG 同定結果ファイル p-value とその順位 G2 群で高発現 M-A plot の A 値と M 値 q-value FDR 閾値判定結果 q-value < 0.05 を満たす DEG が 1 non-deg が 0 G1 群で高発現 2 位も Ras 群 (G2 群 ) で高発現の DEG 53
54 発現変動遺伝子検出結果 TCC を用いた DEG 同定結果ファイル p-value とその順位 G2 群で高発現 M-A plot の A 値と M 値 q-value FDR 閾値判定結果 q-value < 0.05 を満たす DEG が 1 non-deg が 0 G1 群で高発現 3,4 位も Ras 群 (G2 群 ) で高発現の DEG 54
55 発現変動遺伝子検出結果 TCC を用いた DEG 同定結果ファイル p-value とその順位 G2 群で高発現 M-A plot の A 値と M 値 q-value FDR 閾値判定結果 q-value < 0.05 を満たす DEG が 1 non-deg が 0 G1 群で高発現 5 位は Pro 群 (G1 群 ) で高発現の DEG 55
56 発現変動遺伝子検出結果 TCC を用いた DEG 同定結果ファイル p-value とその順位 G2 群で高発現 M-A plot の A 値と M 値 q-value FDR 閾値判定結果 q-value < 0.05 を満たす DEG が 1 non-deg が 0 G1 群で高発現 指定した FDR 閾値 (0.05) をギリギリ満たす 5,669 位の遺伝子 56
57 発現変動遺伝子検出結果 TCC を用いた DEG 同定結果ファイル ハイライトさせたい Gene ID の位置情報を論理値ベクトル obj として取得後 points 関数を用いて obj が TRUE となる要素のみ pch, cex, col オプションを駆使して追加で描画している rcode_srp017142_highlight.txt( の一部 ) G2 群で高発現 G1 群で高発現 57

58 テンプレートとの違いは赤矢印部分のみ rcode_srp017142_highlight.txt( の一部 ) G2 群で高発現 G1 群で高発現 58
59 Contents( 第 4 回 ) 新規転写物同定 ( ゲノム情報を利用 ) 基本的な考え方 Tophat-Cufflinks パイプライン 可視化 ( ゲノムブラウザや Viewer) 発現量推定 ( 遺伝子レベルと転写物レベル ) RPKM の基本的な考え方 計算時間短縮戦略 ( トランスクリプトーム情報のみを利用 ) カウントデータを用いたサンプル間比較解析 イントロ ( カウントデータ取得まで ) サンプル間クラスタリング 発現変動遺伝子検出 分布やモデル 課題 59

 に相当 (2 7.")
60 59,857 genes 教科書 p 分布やモデルのイントロ TCC を用いた DEG 同定 G1 群 G2 群 M-A plot の M 値は倍率変化 (log 比 ) に相当 ( 倍 G2 群で高発現 ) 60
61 DEG 同定結果 :FDR 閾値の違い TCC を用いた DEG 同定 2,314 DEGs (FDR 0.01%) 5,669 DEGs (FDR 5%) 10,053 DEGs (FDR 40%) FDR 閾値を緩めると得られる DEG 数は増える傾向厳しめ FDR 閾値 緩め 61
62 分布やモデル TCC を用いた DEG 同定 2,314 DEGs (FDR 0.01%) 5,669 DEGs (FDR 5%) 10,053 DEGs (FDR 40%) 黒の分布は non-deg の分布に相当 62
63 59,857 genes 分布やモデル 同一群 (G1 群 ) 同一群 (G2 群 ) Pro 群 vs. Ras 群 G1 群 G2 群 Pro_rep1 群 vs. Pro_rep3 群 黒の分布は non-deg の分布に相当 63
64 59,857 genes 分布やモデル 同一群 (G1 群 ) 同一群 (G2 群 ) Pro 群 vs. Ras 群 G1 群 G2 群 Ras_rep2 群 vs. Ras_rep3 群 黒の分布は non-deg の分布に相当 64
65 59,857 genes 分布やモデル 同一群 (G1 群 ) 同一群 (G2 群 ) Pro 群 vs. Ras 群 G1 群 G2 群 Ras_rep1 群 vs. Ras_rep2 群 黒の分布は non-deg の分布に相当 65
66 59,857 genes 分布やモデル 同一群 (G1 群 ) 同一群 (G2 群 ) Pro 群 vs. Ras 群 G1 群 G2 群 G1 群 G2 群 G1 群 G2 群 同一群内のばらつきの分布 (non-deg 分布 ) 以外のものが DEG と判定されるのが統計的手法の結果... 66
67 統計的手法とは 同一群内の遺伝子のばらつきの程度を把握し 帰無仮説に従う分布の全体像を把握しておく ( モデル構築 ) non-deg のばらつきの程度を把握しておくことと同義 実際に比較したい 2 群の遺伝子のばらつきの程度が non-deg 分布のどのあたりに位置するかを評価 同一群内のばらつきの分布 (non- DEG 分布 ) から遠く離れたところに位置するものは 0 に近い p-value 67
 のど真ん中に位置するものは 1 に近い p-value 68")
68 統計的手法とは 同一群内の遺伝子のばらつきの程度を把握し 帰無仮説に従う分布の全体像を把握しておく ( モデル構築 ) non-deg のばらつきの程度を把握しておくことと同義 実際に比較したい 2 群の遺伝子のばらつきの程度が non-deg 分布のどのあたりに位置するかを評価 同一群内のばらつきの分布 (non- DEG 分布 ) のど真ん中に位置するものは 1 に近い p-value 68
 Pro 群 vs.")

69 59,857 genes 倍率変化の結果 同一群 (G1 群 ) 同一群 (G2 群 ) Pro 群 vs. Ras 群 9,233 DEGs G1 群 G2 群 G1 群 G2 群 G1 群 G2 群 2,731 DEGs 6,718 DEGs 3,390 DEGs 同一群内比較でも多数の偽陽性が検出されている... 69
70 59,857 genes 統計的手法 TCC の結果 同一群 (G1 群 ) 同一群 (G2 群 ) Pro 群 vs. Ras 群 5,669 DEGs G1 群 G2 群 G1 群 G2 群 G1 群 G2 群 7 DEGs 5 DEGs 17 DEGs 同一群内比較でも多少の偽陽性が検出されるが許容範囲... 70
71 rcode_srp017142_nondeg.txt 解析したいサンプルの列番号とサンプル数を指定 パッケージのバージョン次第で結果が変わりうるのは確認済み hoge3_fdr.png 17 DEGs hoge3_fc.png 3,390 DEGs 71
72 non-deg G2 で高発現 DEG DEG 課題用シミュレーションデータ data_hypodata_3vs3.txt(2 群間比較用 ) G1 群 :3サンプル G2 群 :3サンプル全部で10,000 行 6 列 最初の2,000 行分が発現変動遺伝子 (DEG) TCC パッケージを用いて 複製あり 2 群間比較を行う non-deg G1 で高発現 G1:3 反復 G2:3 反復 72
73 課題 data_hypodata_3vs3.txt のサンプル間比較解析を行う 1. TCC パッケージを用いた発現変動遺伝子 (DEG) 検出を行い FDR 閾値が および 0.40 を満たす遺伝子数を示せ また このデータセット中の大まかな DEG 数を示すとともにその根拠を簡単に述べよ FDR 閾値 0.05 を満たす遺伝子数 (q-value < 0.05): FDR 閾値 0.20 を満たす遺伝子数 (q-value < 0.20): FDR 閾値 0.40 を満たす遺伝子数 (q-value < 0.40): このデータセット中に含まれる推定 DEG 数 ( 偽物を差し引いた本物の DEG 数 ): 推定した DEG 数の根拠 : 2. 結果の考察 シミュレーションデータ (data_hypodata_3vs3.txt) のサンプル間クラスタリング結果との比較や 実データ (srp017142_count_bowtie.txt) 解析結果との比較など自由に述べてよい 73
74 多重比較問題 :FDR って何? p-value (false positive rate; FPR) 本当は DEG ではないにもかかわらず DEG と判定してしまう確率 全遺伝子に占める non-deg の割合 ( 分母は遺伝子総数 ) 例 :10,000 個の non-deg からなる遺伝子を p-value < 0.05 で検定すると 10, = 500 個程度の non-deg を間違って DEG と判定することに相当 実際の DEG 検出結果が 900 個だった場合 :500 個は偽物で 400 個は本物と判断 実際の DEG 検出結果が 510 個だった場合 :500 個は偽物で 10 個は本物と判断 実際の DEG 検出結果が 500 個以下の場合 : 全て偽物と判断 q-value (false discovery rate: FDR) DEG と判定した中に含まれる non-deg の割合 Benjamini and Hochberg J. Roy. Stat. Soc. B, 57: , 1995 参考 DEG 中に占める non-deg の割合 ( 分母は DEG と判定された数 ) non-deg の期待値を計算できれば p 値でも上位 x 個でも DEG と判定する手段はなんでもよい 以下は 10,000 遺伝子の検定結果での FDR 計算例 p < を満たす DEG 数が 100 個の場合 :FDR = 10, /100 = 0.1 p < 0.01 を満たす DEG 数が 400 個の場合 :FDR = 10, /400 = 0.25 p < 0.05 を満たす DEG 数が 926 個の場合 :FDR = 10, /926 = 0.54 教科書 p
75 多重比較問題 :FDR って何? Benjamini and Hochberg J. Roy. Stat. Soc. B, 57: , 1995 参考 DEG か non-deg かを判定する閾値を決める問題 有意水準 5% というのが p-value < 0.05 に相当 False discovery rate (FDR) 5% というのが q-value < 0.05 に相当 教科書 p 発現変動ランキング結果は不変なので上位 x 個という決め打ちの場合にはこの問題とは無関係 5% の偽物 ( 本当は non-deg だが DEG と判定してしまう誤り ) を許容すると 5,669 遺伝子が DEG とみなせます 5, = 個が理論上偽物だということ 75
 1% というのが q-value < 0.")
76 多重比較問題 :FDR って何? Benjamini and Hochberg J. Roy. Stat. Soc. B, 57: , 1995 参考 DEG か non-deg かを判定する閾値を決める問題 有意水準 5% というのが p-value < 0.05 に相当 False discovery rate (FDR) 1% というのが q-value < 0.01 に相当 教科書 p 発現変動ランキング結果は不変なので上位 x 個という決め打ちの場合にはこの問題とは無関係 1% の偽物 ( 本当は non-deg だが DEG と判定してしまう誤り ) を許容すると 4,189 遺伝子が DEG とみなせます = 個が理論上偽物だということ 76
 5% というのが q-value < 0.05 に相当 教科書 p111-121 発現変動ランキング結果は不変なので上位 x 個という決め打ちの場合にはこの問題とは無関係 有意水準 0.1% で 59,857 遺伝子を検定すると 4,422 個が棄却された (p < 0.")
77 多重比較問題 :FDR って何? Benjamini and Hochberg J. Roy. Stat. Soc. B, 57: , 1995 参考 DEG か non-deg かを判定する閾値を決める問題 有意水準 0.1% というのが p-value < に相当 False discovery rate (FDR) 5% というのが q-value < 0.05 に相当 教科書 p 発現変動ランキング結果は不変なので上位 x 個という決め打ちの場合にはこの問題とは無関係 有意水準 0.1% で 59,857 遺伝子を検定すると 4,422 個が棄却された (p < を満たすものは 59,857 遺伝子中 4,422 個でした ) 77


78 多重比較問題 :FDR って何? Benjamini and Hochberg J. Roy. Stat. Soc. B, 57: , 1995 参考 DEG か non-deg かを判定する閾値を決める問題 有意水準 0.1% というのが p-value < に相当 False discovery rate (FDR) 5% というのが q-value < 0.05 に相当 教科書 p 発現変動ランキング結果は不変なので上位 x 個という決め打ちの場合にはこの問題とは無関係 p 値の定義から 59,857 遺伝子 = 個分の真の non-deg を DEG と判定ミスするのを許容することに相当 p < を満たす 4,422 個の中に占める偽物の割合は /4,422 = と計算することができる これ ( ) が FDR!! 78

79 参考 過去の講義や講演資料の PDF はこちらから取得可能 79



80 まとめ 参考 NGS 解析に必要な全般的な解説記事 講演予定はこちら リンク先などから芋づる式に情報収集 80
機能ゲノム学(第6回)
") トランスクリプトーム解析の今昔 なぜマイクロアレイ? なぜRNA-Seq? 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 Contents トランスクリプトーム解析の概要 各手法の長所 短所 マイクロアレイ
トランスクリプトーム解析の今昔 なぜマイクロアレイ? なぜRNA-Seq? 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 Contents トランスクリプトーム解析の概要 各手法の長所 短所 マイクロアレイ
機能ゲノム学(第6回)
") RNA-Seqデータ解析における正規化法の選択 :RPKM 値でサンプル間比較は危険?! 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 よりよい正規化法とは? その正規化法によって得られたデータを用いて発現変動の度合いでランキングしたときに
RNA-Seqデータ解析における正規化法の選択 :RPKM 値でサンプル間比較は危険?! 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 よりよい正規化法とは? その正規化法によって得られたデータを用いて発現変動の度合いでランキングしたときに
機能ゲノム学(第6回)
") RNA-Seq データ解析リテラシー 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 2009 年ごろの私 次世代シーケンサー (NGS) 解析についての認識 単に短い塩基配列が沢山あるだけでしょ 得られる配列データって
RNA-Seq データ解析リテラシー 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 2009 年ごろの私 次世代シーケンサー (NGS) 解析についての認識 単に短い塩基配列が沢山あるだけでしょ 得られる配列データって
ゲノム情報解析基礎 ~ Rで塩基配列解析 ~
 ネット接続できないヒトも ダブルクリックでローカルに r_seq.html を起動可能です 実習は デスクトップ上にある hoge フォルダの中身が以下の状態を想定して行います (R で ) 塩基配列解析の利用法 : GC 含量計算から発現変動解析まで東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected]
ネット接続できないヒトも ダブルクリックでローカルに r_seq.html を起動可能です 実習は デスクトップ上にある hoge フォルダの中身が以下の状態を想定して行います (R で ) 塩基配列解析の利用法 : GC 含量計算から発現変動解析まで東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected]
Rでゲノム・トランスクリプトーム解析
 R でゲノム トランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 自己紹介 1995 年 3 月 高知工業高等専門学校 工業化学科卒業 1997 年 3 月 東京農工大学 工学部 物質生物工学科卒業
R でゲノム トランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 自己紹介 1995 年 3 月 高知工業高等専門学校 工業化学科卒業 1997 年 3 月 東京農工大学 工学部 物質生物工学科卒業
Rでトランスクリプトーム解析
 R でトランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 自己紹介 1995 年 3 月 高知工業高等専門学校 工業化学科卒業 1997 年 3 月 東京農工大学 工学部 物質生物工学科卒業
R でトランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 自己紹介 1995 年 3 月 高知工業高等専門学校 工業化学科卒業 1997 年 3 月 東京農工大学 工学部 物質生物工学科卒業
NGSデータ解析入門Webセミナー
 NGS データ解析入門 Web セミナー : RNA-Seq 解析編 1 RNA-Seq データ解析の手順 遺伝子発現量測定 シークエンス マッピング サンプル間比較 機能解析など 2 CLC Genomics Workbench 使用ツール シークエンスデータ メタデータのインポート NGS data import Import Metadata クオリティチェック Create Sequencing
NGS データ解析入門 Web セミナー : RNA-Seq 解析編 1 RNA-Seq データ解析の手順 遺伝子発現量測定 シークエンス マッピング サンプル間比較 機能解析など 2 CLC Genomics Workbench 使用ツール シークエンスデータ メタデータのインポート NGS data import Import Metadata クオリティチェック Create Sequencing
機能ゲノム学(第6回)
") RNAseqによる 定 量 的 解 析 とqPCR マイクロアレイなど との 比 較 東 京 大 学 大 学 院 農 学 生 命 科 学 研 究 科 アグリバイオインフォマティクス 教 育 研 究 ユニット 門 田 幸 二 (かどた こうじ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 自 己 紹 介 1995
RNAseqによる 定 量 的 解 析 とqPCR マイクロアレイなど との 比 較 東 京 大 学 大 学 院 農 学 生 命 科 学 研 究 科 アグリバイオインフォマティクス 教 育 研 究 ユニット 門 田 幸 二 (かどた こうじ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 自 己 紹 介 1995
PowerPoint プレゼンテーション
 V1 次世代シークエンサ実習 II 本講義の内容 Reseq 解析 RNA-seq 解析 公開データ取得 クオリティコントロール マッピング 変異検出 公開データ取得 クオリティコントロール マッピング 発現定量 FPKM を算出します 2 R N A - s e q とは メッセンジャー RNA(mRNA) をキャプチャして次世代シーケンサーでシーケンシングする手法 リファレンスがある生物種の場合
V1 次世代シークエンサ実習 II 本講義の内容 Reseq 解析 RNA-seq 解析 公開データ取得 クオリティコントロール マッピング 変異検出 公開データ取得 クオリティコントロール マッピング 発現定量 FPKM を算出します 2 R N A - s e q とは メッセンジャー RNA(mRNA) をキャプチャして次世代シーケンサーでシーケンシングする手法 リファレンスがある生物種の場合
ゲノム情報解析基礎 ~ Rで塩基配列解析 ~
 トランスクリプトーム解析の現況 ~ マイクロアレイ vs. RNA-seq~ 東京大学 大学院農学生命科学研究科 アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected] http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 スライド PDF はウェブから取得可能です 2 ステレオタイプなイメージ
トランスクリプトーム解析の現況 ~ マイクロアレイ vs. RNA-seq~ 東京大学 大学院農学生命科学研究科 アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected] http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 スライド PDF はウェブから取得可能です 2 ステレオタイプなイメージ
IonTorrent RNA-Seq 解析概要 サーモフィッシャーサイエンティフィックライフテクノロジーズジャパンテクニカルサポート The world leader in serving science
 IonTorrent RNA-Seq 解析概要 2017-03 サーモフィッシャーサイエンティフィックライフテクノロジーズジャパンテクニカルサポート The world leader in serving science 資料概要 この資料は IonTorrent シーケンサーで RNA-Seq (WholeTranscriptome mrna ampliseqrna mirna) 解析を実施されるユーザー様向けの内容となっています
IonTorrent RNA-Seq 解析概要 2017-03 サーモフィッシャーサイエンティフィックライフテクノロジーズジャパンテクニカルサポート The world leader in serving science 資料概要 この資料は IonTorrent シーケンサーで RNA-Seq (WholeTranscriptome mrna ampliseqrna mirna) 解析を実施されるユーザー様向けの内容となっています
シーケンサー利用技術講習会 第10回 サンプルQC、RNAseqライブラリー作製/データ解析実習講習会
 シーケンサー利用技術講習会 第 10 回サンプル QC RNAseq ライブ ラリー作製 / データ解析実習講習会 理化学研究所ライフサイエンス技術基盤研究センターゲノムネットワーク解析支援施設田上道平 次世代シーケンサー Sequencer File Format Output(Max) Read length Illumina Hiseq2500 Fastq 600 Gb 100 bp Life
シーケンサー利用技術講習会 第 10 回サンプル QC RNAseq ライブ ラリー作製 / データ解析実習講習会 理化学研究所ライフサイエンス技術基盤研究センターゲノムネットワーク解析支援施設田上道平 次世代シーケンサー Sequencer File Format Output(Max) Read length Illumina Hiseq2500 Fastq 600 Gb 100 bp Life
ChIP-seq
 ChIP-seq 1 ChIP-seq 解析原理 ChIP サンプルのフラグメントでは タンパク質結合部位付近にそれぞれ Forward と Reverse のリードがマップされることが予想される ChIP のサンプルでは Forward と Reverse のリードを 3 側へシフトさせ ChIP のピークを算出する コントロールサンプルでは ChIP のサンプルとは異なり 特定の場所に多くマップされないため
ChIP-seq 1 ChIP-seq 解析原理 ChIP サンプルのフラグメントでは タンパク質結合部位付近にそれぞれ Forward と Reverse のリードがマップされることが予想される ChIP のサンプルでは Forward と Reverse のリードを 3 側へシフトさせ ChIP のピークを算出する コントロールサンプルでは ChIP のサンプルとは異なり 特定の場所に多くマップされないため
Microsoft Word - 1 color Normalization Document _Agilent version_ .doc
 color 実験の Normalization color 実験で得られた複数のアレイデータを相互比較するためには Normalization( 正規化 ) が必要です 2 つのサンプルを異なる色素でラベル化し 競合ハイブリダイゼーションさせる 2color 実験では 基本的に Dye Normalization( 色素補正 ) が適用されますが color 実験では データの特徴と実験の目的 (
color 実験の Normalization color 実験で得られた複数のアレイデータを相互比較するためには Normalization( 正規化 ) が必要です 2 つのサンプルを異なる色素でラベル化し 競合ハイブリダイゼーションさせる 2color 実験では 基本的に Dye Normalization( 色素補正 ) が適用されますが color 実験では データの特徴と実験の目的 (
データ科学2.pptx
 データ科学 多重検定 2 mul%ple test False Discovery Rate 藤博幸 前回の復習 1 多くの検定を繰り返す時には 単純に個々の検定を繰り返すだけでは不十分 5% 有意水準ということは, 1000 回検定を繰り返すと, 50 回くらいは帰無仮説が正しいのに 間違って棄却されてすまうじちがあるということ ex) 1 万個の遺伝子について 正常細胞とガン細胞で それぞれの遺伝子の発現に差があるかどうかを検定
データ科学 多重検定 2 mul%ple test False Discovery Rate 藤博幸 前回の復習 1 多くの検定を繰り返す時には 単純に個々の検定を繰り返すだけでは不十分 5% 有意水準ということは, 1000 回検定を繰り返すと, 50 回くらいは帰無仮説が正しいのに 間違って棄却されてすまうじちがあるということ ex) 1 万個の遺伝子について 正常細胞とガン細胞で それぞれの遺伝子の発現に差があるかどうかを検定
GWB_RNA-Seq_
 CLC Genomics Workbench ウェブトレーニングセミナー : RNA-Seq 編 フィルジェン株式会社バイオサイエンス部 ([email protected]) 1 Advanced RNA-Seq プラグイン CLC Genomics Workbench 9.0 / Biomedical Genomics Workbench 3.0 以降で使用可能な無償プラグイン RNA-Seq
CLC Genomics Workbench ウェブトレーニングセミナー : RNA-Seq 編 フィルジェン株式会社バイオサイエンス部 ([email protected]) 1 Advanced RNA-Seq プラグイン CLC Genomics Workbench 9.0 / Biomedical Genomics Workbench 3.0 以降で使用可能な無償プラグイン RNA-Seq
機能ゲノム学(第6回)
") R でトランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 自己紹介 2002 年 3 月 東京大学 大学院農学生命科学研究科博士課程修了 学位論文 : cdna マイクロアレイを用いた遺伝子発現解析手法の開発
R でトランスクリプトーム解析 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 自己紹介 2002 年 3 月 東京大学 大学院農学生命科学研究科博士課程修了 学位論文 : cdna マイクロアレイを用いた遺伝子発現解析手法の開発
RNA-seq
 RNA-seq 1 RNA-seq 解析フロー RNA-seq インポート クオリティチェック RNA-seq 発現差解析 この資料では RNA-seq からの説明となりますが インポート クオリティチェックについては サポート資料のページより内容をご確認いただけます 2 データ 発現解析用デモデータは 以下よりダウンロードいただけます ES 細胞 (ESC) と神経前駆細胞 (NPC) の発現解析を小さなデモデータで行えます
RNA-seq 1 RNA-seq 解析フロー RNA-seq インポート クオリティチェック RNA-seq 発現差解析 この資料では RNA-seq からの説明となりますが インポート クオリティチェックについては サポート資料のページより内容をご確認いただけます 2 データ 発現解析用デモデータは 以下よりダウンロードいただけます ES 細胞 (ESC) と神経前駆細胞 (NPC) の発現解析を小さなデモデータで行えます
特論I
 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 2 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 [email protected] 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG 概論 現状や展望など 講義のみ 第 2 回 (2014
講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 2 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 [email protected] 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG 概論 現状や展望など 講義のみ 第 2 回 (2014
CLC Genomics Workbench ウェブトレーニングセミナー: 変異解析編
 CLC Genomics Workbench ウェブトレーニングセミナー : 遺伝子発現解析編 12 th Feb., 2016 フィルジェン株式会社バイオサイエンス部 [email protected] Feb., 2016_V2 1 遺伝子発現解析概要 本日のセミナーにおける解析の流れ及び使用するツール名 ( 図中赤枠部分 ) Case Control インポート インポート インポート
CLC Genomics Workbench ウェブトレーニングセミナー : 遺伝子発現解析編 12 th Feb., 2016 フィルジェン株式会社バイオサイエンス部 [email protected] Feb., 2016_V2 1 遺伝子発現解析概要 本日のセミナーにおける解析の流れ及び使用するツール名 ( 図中赤枠部分 ) Case Control インポート インポート インポート
基本的な利用法
 (R で ) 塩基配列解析 基本的な利用法 Macintosh 版 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける 1. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール 2. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握
(R で ) 塩基配列解析 基本的な利用法 Macintosh 版 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける 1. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール 2. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握
特論I
 2016.02.01 版 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 3 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 [email protected] Jun 25, 2014 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG
2016.02.01 版 講義室後ろにある USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 農学生命情報科学特論 I 第 3 回 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 [email protected] Jun 25, 2014 1 講義予定 第 1 回 (2014 年 6 月 11 日 ) 西 :NSG
PrimerArray® Analysis Tool Ver.2.2
 研究用 PrimerArray Analysis Tool Ver.2.2 説明書 v201801 PrimerArray Analysis Tool Ver.2.2 は PrimerArray( 製品コード PH001 ~ PH007 PH009 ~ PH015 PN001 ~ PN015) で得られたデータを解析するためのツールで コントロールサンプルと 1 種類の未知サンプル間の比較が可能です
研究用 PrimerArray Analysis Tool Ver.2.2 説明書 v201801 PrimerArray Analysis Tool Ver.2.2 は PrimerArray( 製品コード PH001 ~ PH007 PH009 ~ PH015 PN001 ~ PN015) で得られたデータを解析するためのツールで コントロールサンプルと 1 種類の未知サンプル間の比較が可能です
機能ゲノム学
 08.05.08 版 講義資料 PDF が講義のページからダウンロード可能です 講義資料の印刷物はありません 課題用の A4 一枚はあります 第 回出席予定の持込み PC の方は 当日までに Java のインストールをしておいてください 機能ゲノム学第 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム 微生物科学イノベーション連携研究機構門田幸二 ( かどたこうじ ) [email protected]
08.05.08 版 講義資料 PDF が講義のページからダウンロード可能です 講義資料の印刷物はありません 課題用の A4 一枚はあります 第 回出席予定の持込み PC の方は 当日までに Java のインストールをしておいてください 機能ゲノム学第 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム 微生物科学イノベーション連携研究機構門田幸二 ( かどたこうじ ) [email protected]
PowerPoint Presentation
 エピジェノミクス解析編 2016/08/10 Filgen ChIP-seq (Transfactor & Histone), Bisulfite webex seminar 株式会社キアゲンアプライドアドバンストゲノミクス宮本真理, PhD 1 アジェンダ ChIP-seq 解析 Transcription Factor ChIP-seq Histone ChIP-seq Bisulfite-seq
エピジェノミクス解析編 2016/08/10 Filgen ChIP-seq (Transfactor & Histone), Bisulfite webex seminar 株式会社キアゲンアプライドアドバンストゲノミクス宮本真理, PhD 1 アジェンダ ChIP-seq 解析 Transcription Factor ChIP-seq Histone ChIP-seq Bisulfite-seq
NGS速習コース
 バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-3. R 各種パッケージ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) [email protected] http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-3. R 各種パッケージ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) [email protected] http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
農学生命情報科学特論I
 2015.07.01 版 USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 前回 (6/23) の hoge フォルダがデスクトップに残っているかもしれないのでご注意ください 農学生命情報科学 特論 I 第 3 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected]
2015.07.01 版 USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください 前回 (6/23) の hoge フォルダがデスクトップに残っているかもしれないのでご注意ください 農学生命情報科学 特論 I 第 3 回 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected]
GWB
 NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 CLC Genomics Workbench 使用ツール シークエンスデータのインポート NGS data import クオリティチェック QC for Sequencing Reads Trim Reads 参照ゲノム配列へのマッピング 再アライメント
NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 CLC Genomics Workbench 使用ツール シークエンスデータのインポート NGS data import クオリティチェック QC for Sequencing Reads Trim Reads 参照ゲノム配列へのマッピング 再アライメント
GWB
 NGS データ解析入門 Web セミナー : De Novo シークエンス解析編 1 NGS 新規ゲノム配列解析の手順 シークエンス 遺伝子領域の検出 アセンブル データベース検索 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 前処理 コンティグ配列の作成 CLC Genomics Workbench 遺伝子領域の検出 Blast2GO PRO データベース検索
NGS データ解析入門 Web セミナー : De Novo シークエンス解析編 1 NGS 新規ゲノム配列解析の手順 シークエンス 遺伝子領域の検出 アセンブル データベース検索 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 前処理 コンティグ配列の作成 CLC Genomics Workbench 遺伝子領域の検出 Blast2GO PRO データベース検索
Rでゲノム・トランスクリプトーム解析
 06.03.05 版 実習用 PC のデスクトップ上に hoge フォルダがあります この中に解析に必要な入力ファイルがあります ネットワーク不具合時は ローカル環境で html ファイルを起動して各自対応してください R で塩基配列解析 : ゲノム解析からトランスクリプトーム解析まで 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ )
06.03.05 版 実習用 PC のデスクトップ上に hoge フォルダがあります この中に解析に必要な入力ファイルがあります ネットワーク不具合時は ローカル環境で html ファイルを起動して各自対応してください R で塩基配列解析 : ゲノム解析からトランスクリプトーム解析まで 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ )
NGSハンズオン講習会
 207.08.08 版 プラスアルファの内容です NGS 解析 ( 初 ~ 中級 ) ゲノムアセンブリ後の各種解析の補足資料 ( プラスアルファ ) 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム寺田朋子 門田幸二 Aug 29-30 207 Contents Gepard でドットプロット 連載第 8 回 W5-3 で最も長い sequence 同士のドットプロットを実行できなかったが
207.08.08 版 プラスアルファの内容です NGS 解析 ( 初 ~ 中級 ) ゲノムアセンブリ後の各種解析の補足資料 ( プラスアルファ ) 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム寺田朋子 門田幸二 Aug 29-30 207 Contents Gepard でドットプロット 連載第 8 回 W5-3 で最も長い sequence 同士のドットプロットを実行できなかったが
Qlucore_seminar_slide_180604
 シングルセル RNA-Seq のための 情報解析 フィルジェン株式会社バイオサイエンス部 ([email protected]) 1 シングルセル RNA-Seq シングルセル RNA-Seq のデータ解析では 通常の RNA-Seq データの解析手法に加え データセット内の各細胞の遺伝子発現プロファイルの違いを俯瞰できるような 強力な情報解析アルゴリズムと データのビジュアライズ機能を利用する必要がある
シングルセル RNA-Seq のための 情報解析 フィルジェン株式会社バイオサイエンス部 ([email protected]) 1 シングルセル RNA-Seq シングルセル RNA-Seq のデータ解析では 通常の RNA-Seq データの解析手法に加え データセット内の各細胞の遺伝子発現プロファイルの違いを俯瞰できるような 強力な情報解析アルゴリズムと データのビジュアライズ機能を利用する必要がある
GWB
 NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 参照ゲノム配列へのマッピング 再アライメント 変異検出 CLC Genomics Workbench または Biomedical Genomics Workbench
NGS データ解析入門 Web セミナー : 変異解析編 1 NGS 変異データ解析の手順 シークエンス 変異検出 マッピング データの精査 解釈 2 解析ワークフローと使用ソフトウェア シークエンスデータのインポート クオリティチェック 参照ゲノム配列へのマッピング 再アライメント 変異検出 CLC Genomics Workbench または Biomedical Genomics Workbench
Rインストール手順
 R 本体は最新のリリース版 R パッケージは 必要最小限プラスアルファ の推奨インストール手順を示します R 本体とパッケージのインストール Windows 版 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected] http://www.iu.a.u-tokyo.ac.jp/~kadota/
R 本体は最新のリリース版 R パッケージは 必要最小限プラスアルファ の推奨インストール手順を示します R 本体とパッケージのインストール Windows 版 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected] http://www.iu.a.u-tokyo.ac.jp/~kadota/
リード・ゲノム・アノテーションインポート
 リード ゲノム アノテーションインポート 1 Location と Folder ロケーション フォルダ Genomics Workbenchではデータを以下のような階層構造で保存可能です フォルダの一番上位の階層を Location と呼び その下の階層を Folder と呼びます データの保存場所はロケーション毎に設定可能です たとえばあるデータは C ドライブに保存し あるデータは D ドライブに保存するといった事が可能です
リード ゲノム アノテーションインポート 1 Location と Folder ロケーション フォルダ Genomics Workbenchではデータを以下のような階層構造で保存可能です フォルダの一番上位の階層を Location と呼び その下の階層を Folder と呼びます データの保存場所はロケーション毎に設定可能です たとえばあるデータは C ドライブに保存し あるデータは D ドライブに保存するといった事が可能です
ビジネス統計 統計基礎とエクセル分析 正誤表
 ビジネス統計統計基礎とエクセル分析 ビジネス統計スペシャリスト エクセル分析スペシャリスト 公式テキスト正誤表と学習用データ更新履歴 平成 30 年 5 月 14 日現在 公式テキスト正誤表 頁場所誤正修正 6 知識編第 章 -3-3 最頻値の解説内容 たとえば, 表.1 のデータであれば, 最頻値は 167.5cm というたとえば, 表.1 のデータであれば, 最頻値は 165.0cm ということになります
ビジネス統計統計基礎とエクセル分析 ビジネス統計スペシャリスト エクセル分析スペシャリスト 公式テキスト正誤表と学習用データ更新履歴 平成 30 年 5 月 14 日現在 公式テキスト正誤表 頁場所誤正修正 6 知識編第 章 -3-3 最頻値の解説内容 たとえば, 表.1 のデータであれば, 最頻値は 167.5cm というたとえば, 表.1 のデータであれば, 最頻値は 165.0cm ということになります
Kumamoto University Center for Multimedia and Information Technologies Lab. 熊本大学アプリケーション実験 ~ 実環境における無線 LAN 受信電波強度を用いた位置推定手法の検討 ~ InKIAI 宮崎県美郷
 熊本大学アプリケーション実験 ~ 実環境における無線 LAN 受信電波強度を用いた位置推定手法の検討 ~ InKIAI プロジェクト @ 宮崎県美郷町 熊本大学副島慶人川村諒 1 実験の目的 従来 信号の受信電波強度 (RSSI:RecevedSgnal StrengthIndcator) により 対象の位置を推定する手法として 無線 LAN の AP(AccessPont) から受信する信号の減衰量をもとに位置を推定する手法が多く検討されている
熊本大学アプリケーション実験 ~ 実環境における無線 LAN 受信電波強度を用いた位置推定手法の検討 ~ InKIAI プロジェクト @ 宮崎県美郷町 熊本大学副島慶人川村諒 1 実験の目的 従来 信号の受信電波強度 (RSSI:RecevedSgnal StrengthIndcator) により 対象の位置を推定する手法として 無線 LAN の AP(AccessPont) から受信する信号の減衰量をもとに位置を推定する手法が多く検討されている
Agilent 1色法 2条件比較 繰り返し実験なし
 GeneSpring GX11.0.2 ビギナーズガイド Agilent 1 色法 2 条件の比較繰り返し実験あり 適用 薬剤非投与と投与の解析 Wild type と Knock out の解析 正常細胞と病態細胞の解析 など ビギナーズガイドは 様々なマイクロアレイの実験デザインがあるなかで 実験デザインの種類ごとに適切なデータ解析の流れを 実例とともに紹介するガイドブックです ご自分の実験デザインに適合したガイドをお使いください
GeneSpring GX11.0.2 ビギナーズガイド Agilent 1 色法 2 条件の比較繰り返し実験あり 適用 薬剤非投与と投与の解析 Wild type と Knock out の解析 正常細胞と病態細胞の解析 など ビギナーズガイドは 様々なマイクロアレイの実験デザインがあるなかで 実験デザインの種類ごとに適切なデータ解析の流れを 実例とともに紹介するガイドブックです ご自分の実験デザインに適合したガイドをお使いください
PowerPoint プレゼンテーション
 バイオインフォマティクス 講習会 V 事前準備 が完了されている方は コンテナの起動 ファイルのコピー (Windows) まで 進めておいてください メニュー 1. 環境構築の確認 2. 基本的なLinuxコマンド 3. ツールのインストール 4. NGSデータの基礎知識と前処理 5. トランスクリプトのアッセンブル 6. RNA-seqのリファレンスcDNAマッピングとFPKM 算出 7. RNA-seqのリファレンスゲノムマッピングとFPKM
バイオインフォマティクス 講習会 V 事前準備 が完了されている方は コンテナの起動 ファイルのコピー (Windows) まで 進めておいてください メニュー 1. 環境構築の確認 2. 基本的なLinuxコマンド 3. ツールのインストール 4. NGSデータの基礎知識と前処理 5. トランスクリプトのアッセンブル 6. RNA-seqのリファレンスcDNAマッピングとFPKM 算出 7. RNA-seqのリファレンスゲノムマッピングとFPKM
次世代シークエンサーを用いたがんクリニカルシークエンス解析
 次世代シークエンサーを用いた がんクリニカルシークエンス解析 フィルジェン株式会社バイオサイエンス部 ([email protected]) 1 がん遺伝子パネル がん関連遺伝子のターゲットシークエンス用のアッセイキット コストの低減や 研究プログラムの簡素化に有用 網羅的シークエンス解析の場合に比べて 1 遺伝子あたりのシークエンス量が増えるため より高感度な変異の検出が可能 2 変異データ解析パイプライン
次世代シークエンサーを用いた がんクリニカルシークエンス解析 フィルジェン株式会社バイオサイエンス部 ([email protected]) 1 がん遺伝子パネル がん関連遺伝子のターゲットシークエンス用のアッセイキット コストの低減や 研究プログラムの簡素化に有用 網羅的シークエンス解析の場合に比べて 1 遺伝子あたりのシークエンス量が増えるため より高感度な変異の検出が可能 2 変異データ解析パイプライン
統計的データ解析
 統計的データ解析 011 011.11.9 林田清 ( 大阪大学大学院理学研究科 ) 連続確率分布の平均値 分散 比較のため P(c ) c 分布 自由度 の ( カイ c 平均値 0, 標準偏差 1の正規分布 に従う変数 xの自乗和 c x =1 が従う分布を自由度 の分布と呼ぶ 一般に自由度の分布は f /1 c / / ( c ) {( c ) e }/ ( / ) 期待値 二乗 ) 分布 c
統計的データ解析 011 011.11.9 林田清 ( 大阪大学大学院理学研究科 ) 連続確率分布の平均値 分散 比較のため P(c ) c 分布 自由度 の ( カイ c 平均値 0, 標準偏差 1の正規分布 に従う変数 xの自乗和 c x =1 が従う分布を自由度 の分布と呼ぶ 一般に自由度の分布は f /1 c / / ( c ) {( c ) e }/ ( / ) 期待値 二乗 ) 分布 c
機能ゲノム学(第6回)
") バイオインフォマティクス次世代シーケンサー (NGS) 編 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 バイオインフォマティクス人材育成講座 スタンダードコース 2 自己紹介 1995 年 3 月 高知工業高等専門学校
バイオインフォマティクス次世代シーケンサー (NGS) 編 東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) http://www.iu.a.u-tokyo.ac.jp/~kadota/ [email protected] 1 バイオインフォマティクス人材育成講座 スタンダードコース 2 自己紹介 1995 年 3 月 高知工業高等専門学校
Excelによる統計分析検定_知識編_小塚明_5_9章.indd
 第7章57766 検定と推定 サンプリングによって得られた標本から, 母集団の統計的性質に対して推測を行うことを統計的推測といいます 本章では, 推測統計の根幹をなす仮説検定と推定の基本的な考え方について説明します 前章までの知識を用いて, 具体的な分析を行います 本章以降の知識は操作編での操作に直接関連していますので, 少し聞きなれない言葉ですが, 帰無仮説 有意水準 棄却域 などの意味を理解して,
第7章57766 検定と推定 サンプリングによって得られた標本から, 母集団の統計的性質に対して推測を行うことを統計的推測といいます 本章では, 推測統計の根幹をなす仮説検定と推定の基本的な考え方について説明します 前章までの知識を用いて, 具体的な分析を行います 本章以降の知識は操作編での操作に直接関連していますので, 少し聞きなれない言葉ですが, 帰無仮説 有意水準 棄却域 などの意味を理解して,
NGSハンズオン講習会
 205.07.27 版 配布する USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください NGS ハンズオン 講習会 :R 基礎 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected] http://www.iu.a.u-tokyo.ac.jp/~kadota/ Contents(
205.07.27 版 配布する USB メモリ中の hoge フォルダをデスクトップにコピーしておいてください NGS ハンズオン 講習会 :R 基礎 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected] http://www.iu.a.u-tokyo.ac.jp/~kadota/ Contents(
Microsoft Word - å“Ÿåłžå¸°173.docx
 回帰分析 ( その 3) 経済情報処理 価格弾力性の推定ある商品について その購入量を w 単価を p とし それぞれの変化量を w p で表 w w すことにする この時 この商品の価格弾力性 は により定義される これ p p は p が 1 パーセント変化した場合に w が何パーセント変化するかを示したものである ここで p を 0 に近づけていった極限を考えると d ln w 1 dw dw
回帰分析 ( その 3) 経済情報処理 価格弾力性の推定ある商品について その購入量を w 単価を p とし それぞれの変化量を w p で表 w w すことにする この時 この商品の価格弾力性 は により定義される これ p p は p が 1 パーセント変化した場合に w が何パーセント変化するかを示したものである ここで p を 0 に近づけていった極限を考えると d ln w 1 dw dw
RNA-seq
 CLC Genomics Workbench ハンズオントレーニング RNA-seq 株式会社 CLCバイオジャパンシニアフィールドバイオインフォマティクスサイエンティスト宮本真理 Ph.D. [email protected] 1 [email protected] 2 アジェンダ Genomics Workbench 概要 今日のデータ RNA-seq 解析 データインポート QC
CLC Genomics Workbench ハンズオントレーニング RNA-seq 株式会社 CLCバイオジャパンシニアフィールドバイオインフォマティクスサイエンティスト宮本真理 Ph.D. [email protected] 1 [email protected] 2 アジェンダ Genomics Workbench 概要 今日のデータ RNA-seq 解析 データインポート QC
Partek Flow リリースノート バージョン : Partek Flow バージョン は高速化と使い勝手の改善のための新機能やパフォーマンス向上を含んでいます このバージョンへアップグレードするためには Partek Flow インストールガイド
 Partek Flow リリースノート バージョン : 5.0.16.0414 Partek Flow バージョン 5.0.16.0414 は高速化と使い勝手の改善のための新機能やパフォーマンス向上を含んでいます このバージョンへアップグレードするためには Partek Flow インストールガイド内のインストール手順を実行して下さい 改善点を以下に列挙します Partek Flow ホームページ
Partek Flow リリースノート バージョン : 5.0.16.0414 Partek Flow バージョン 5.0.16.0414 は高速化と使い勝手の改善のための新機能やパフォーマンス向上を含んでいます このバージョンへアップグレードするためには Partek Flow インストールガイド内のインストール手順を実行して下さい 改善点を以下に列挙します Partek Flow ホームページ
NGS速習コース
 バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-4. R Bioconductor I 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) [email protected] http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
バイオインフォマティクス人材育成カリキュラム ( 次世代シークエンサ ) 速習コース 3. データ解析基礎 3-4. R Bioconductor I 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット門田幸二 ( かどたこうじ ) [email protected] http://www.iu.a.u-tokyo.ac.jp/~kadota/ 1 Contents
配付資料 自習用テキスト 解析サンプル配布ページ 2
 分子系統樹推定法 理論と応用 2009年11月6日 筑波大 院 生命環境 田辺晶史 配付資料 自習用テキスト 解析サンプル配布ページ http://www.fifthdimension.jp/documents/molphytextbook/ 2 参考書籍 分子系統学 3 参考書籍 統計的モデル選択とベイジアンMCMC 4 祖先的な形質 問題 OTU左の の色は表現型形質の状態を表している 赤と青
分子系統樹推定法 理論と応用 2009年11月6日 筑波大 院 生命環境 田辺晶史 配付資料 自習用テキスト 解析サンプル配布ページ http://www.fifthdimension.jp/documents/molphytextbook/ 2 参考書籍 分子系統学 3 参考書籍 統計的モデル選択とベイジアンMCMC 4 祖先的な形質 問題 OTU左の の色は表現型形質の状態を表している 赤と青
KEGG.ppt
 1 2 3 4 KEGG: Kyoto Encyclopedia of Genes and Genomes http://www.genome.jp/kegg/kegg2.html http://www.genome.jp/kegg/kegg_ja.html 5 KEGG PATHWAY 生体内(外)の分子間ネットワーク図 代謝系 12カテゴリ 中間代謝 二次代謝 薬の 代謝 全体像 制御系 20カテゴリ
1 2 3 4 KEGG: Kyoto Encyclopedia of Genes and Genomes http://www.genome.jp/kegg/kegg2.html http://www.genome.jp/kegg/kegg_ja.html 5 KEGG PATHWAY 生体内(外)の分子間ネットワーク図 代謝系 12カテゴリ 中間代謝 二次代謝 薬の 代謝 全体像 制御系 20カテゴリ
免疫形式文法
 遺伝子発現解析入門 中岡慎治 目次 はじめに 遺伝子発現 ( トランスクリプトーム ) 解析とはマイクロアレイ (MA) の原理と応用途次世代シーケンサー (NGS) の原理と応用途 [ 補足 ] 次世代シーケンサーの活用事例 [metagenome/chip-seq] etc 遺伝子発現解析の統計手法 正規化の必要性と手法 [MA/NGS] 発現変動解析 (Differential Expressed
遺伝子発現解析入門 中岡慎治 目次 はじめに 遺伝子発現 ( トランスクリプトーム ) 解析とはマイクロアレイ (MA) の原理と応用途次世代シーケンサー (NGS) の原理と応用途 [ 補足 ] 次世代シーケンサーの活用事例 [metagenome/chip-seq] etc 遺伝子発現解析の統計手法 正規化の必要性と手法 [MA/NGS] 発現変動解析 (Differential Expressed
バイオインフォマティクスⅠ
 バイオインフォマティクス ( 第 5 回 ) 慶應義塾大学生命情報学科 榊原康文 多重アライメントの解 0 2 3 4 5 6 7 j Q T S Y T R Y Q T - Y T R K 0 0-9 -20-44 -52-63 -72-90 Q -6 2 0-6 -4-25 -34-52 2 S -32 5 30 4 6-5 -4-32 3 Y -48-4 2 38 27 8 0 4 P -64-27
バイオインフォマティクス ( 第 5 回 ) 慶應義塾大学生命情報学科 榊原康文 多重アライメントの解 0 2 3 4 5 6 7 j Q T S Y T R Y Q T - Y T R K 0 0-9 -20-44 -52-63 -72-90 Q -6 2 0-6 -4-25 -34-52 2 S -32 5 30 4 6-5 -4-32 3 Y -48-4 2 38 27 8 0 4 P -64-27
サンプルのマルチプレックスおよび下流の解析におけるインデックスのミスアサインメントの影響
 サンプルのマルチプレックスおよび下流の解析におけるインデックスのミスアサインメントの影響 インデックスのミスアサインメントの原因と インデックスホッピングの影響を軽減するベストプラクティス はじめに 次世代シーケンス (NGS) 技術の改良により シーケンススピードが大幅に向上し データ出力が飛躍的に増加したことで 現在のシーケンスプラットフォームにおいて大規模なサンプルの解析が可能になりました 10
サンプルのマルチプレックスおよび下流の解析におけるインデックスのミスアサインメントの影響 インデックスのミスアサインメントの原因と インデックスホッピングの影響を軽減するベストプラクティス はじめに 次世代シーケンス (NGS) 技術の改良により シーケンススピードが大幅に向上し データ出力が飛躍的に増加したことで 現在のシーケンスプラットフォームにおいて大規模なサンプルの解析が可能になりました 10
Microsoft PowerPoint _webinar_RNAExpress.erikibukawa_配布用.pptx
 2014 年 10 月 17 日イルミナサポートウェビナー RNA Seq を始めよう! BaseSpace で行う かんたん NGS データ解析 < RNA Express > イルミナ株式会社バイオインフォマティクスサポートサイエンティスト癸生川絵里 (Eri Kibukawa) 2013 2014 Illumina, Inc. All rights reserved. Illumina, 24sure,
2014 年 10 月 17 日イルミナサポートウェビナー RNA Seq を始めよう! BaseSpace で行う かんたん NGS データ解析 < RNA Express > イルミナ株式会社バイオインフォマティクスサポートサイエンティスト癸生川絵里 (Eri Kibukawa) 2013 2014 Illumina, Inc. All rights reserved. Illumina, 24sure,
青焼 1章[15-52].indd
![青焼 1章[15-52].indd](/thumbs/86/94313777.jpg "青焼 1章[15-52].indd") 1 第 1 章統計の基礎知識 1 1 なぜ統計解析が必要なのか? 人間は自分自身の経験にもとづいて 感覚的にものごとを判断しがちである 例えばある疾患に対する標準治療薬の有効率が 50% であったとする そこに新薬が登場し ある医師がその新薬を 5 人の患者に使ったところ 4 人が有効と判定されたとしたら 多くの医師はこれまでの標準治療薬よりも新薬のほうが有効性が高そうだと感じることだろう しかし
1 第 1 章統計の基礎知識 1 1 なぜ統計解析が必要なのか? 人間は自分自身の経験にもとづいて 感覚的にものごとを判断しがちである 例えばある疾患に対する標準治療薬の有効率が 50% であったとする そこに新薬が登場し ある医師がその新薬を 5 人の患者に使ったところ 4 人が有効と判定されたとしたら 多くの医師はこれまでの標準治療薬よりも新薬のほうが有効性が高そうだと感じることだろう しかし
EBNと疫学
 推定と検定 57 ( 復習 ) 記述統計と推測統計 統計解析は大きく 2 つに分けられる 記述統計 推測統計 記述統計 観察集団の特性を示すもの 代表値 ( 平均値や中央値 ) や ばらつきの指標 ( 標準偏差など ) 図表を効果的に使う 推測統計 観察集団のデータから母集団の特性を 推定 する 平均 / 分散 / 係数値などの推定 ( 点推定 ) 点推定値のばらつきを調べる ( 区間推定 ) 検定統計量を用いた検定
推定と検定 57 ( 復習 ) 記述統計と推測統計 統計解析は大きく 2 つに分けられる 記述統計 推測統計 記述統計 観察集団の特性を示すもの 代表値 ( 平均値や中央値 ) や ばらつきの指標 ( 標準偏差など ) 図表を効果的に使う 推測統計 観察集団のデータから母集団の特性を 推定 する 平均 / 分散 / 係数値などの推定 ( 点推定 ) 点推定値のばらつきを調べる ( 区間推定 ) 検定統計量を用いた検定
141025mishima
 NGS (RNAseq) »NGS Now Generation Sequencer»NGS»» 4 NGS(Next Generation Sequencer) Now Generation Sequencer http://www.youtube.com/watch?v=womkfikwlxm http://www.youtube.com/watch?v=mxkya9xcvbq http://www.youtube.com/watch?v=nhcj8ptycfc
NGS (RNAseq) »NGS Now Generation Sequencer»NGS»» 4 NGS(Next Generation Sequencer) Now Generation Sequencer http://www.youtube.com/watch?v=womkfikwlxm http://www.youtube.com/watch?v=mxkya9xcvbq http://www.youtube.com/watch?v=nhcj8ptycfc
Microsoft PowerPoint - DigitalMedia2_3b.pptx
 Contents デジタルメディア処理 2 の概要 フーリエ級数展開と 離散とその性質 周波数フィルタリング 担当 : 井尻敬 とは ( ) FourierSound.py とは ( ) FourierSound.py 横軸が時間の関数を 横軸が周波数の関数に変換する 法 声周波数 周波数 ( 係数番号 ) 後の関数は元信号に含まれる正弦波の量を す 中央に近いほど低周波, 外ほどが 周波 中央 (
Contents デジタルメディア処理 2 の概要 フーリエ級数展開と 離散とその性質 周波数フィルタリング 担当 : 井尻敬 とは ( ) FourierSound.py とは ( ) FourierSound.py 横軸が時間の関数を 横軸が周波数の関数に変換する 法 声周波数 周波数 ( 係数番号 ) 後の関数は元信号に含まれる正弦波の量を す 中央に近いほど低周波, 外ほどが 周波 中央 (
AJACS18_ ppt
 1, 1, 1, 1, 1, 1,2, 1,2, 1 1 DDBJ 2 AJACS3 2010 6 414:20-15:20 2231 DDBJ DDBJ DDBJ DDBJ NCBI (GenBank) DDBJ EBI (EMBL-Bank) GEO DDBJ Omics ARchive(DOR) ArrayExpress DTA (DDBJ Trace Archive) DRA (DDBJ
1, 1, 1, 1, 1, 1,2, 1,2, 1 1 DDBJ 2 AJACS3 2010 6 414:20-15:20 2231 DDBJ DDBJ DDBJ DDBJ NCBI (GenBank) DDBJ EBI (EMBL-Bank) GEO DDBJ Omics ARchive(DOR) ArrayExpress DTA (DDBJ Trace Archive) DRA (DDBJ
多変量解析 ~ 重回帰分析 ~ 2006 年 4 月 21 日 ( 金 ) 南慶典
 南慶典") 多変量解析 ~ 重回帰分析 ~ 2006 年 4 月 21 日 ( 金 ) 南慶典 重回帰分析とは? 重回帰分析とは複数の説明変数から目的変数との関係性を予測 評価説明変数 ( 数量データ ) は目的変数を説明するのに有効であるか得られた関係性より未知のデータの妥当性を判断する これを重回帰分析という つまり どんなことをするのか? 1 最小 2 乗法により重回帰モデルを想定 2 自由度調整済寄与率を求め
多変量解析 ~ 重回帰分析 ~ 2006 年 4 月 21 日 ( 金 ) 南慶典 重回帰分析とは? 重回帰分析とは複数の説明変数から目的変数との関係性を予測 評価説明変数 ( 数量データ ) は目的変数を説明するのに有効であるか得られた関係性より未知のデータの妥当性を判断する これを重回帰分析という つまり どんなことをするのか? 1 最小 2 乗法により重回帰モデルを想定 2 自由度調整済寄与率を求め
基本的な利用法
 到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握 ) 4. ありがちなミス のところで示しているエラーメッセージとその原因をきっちり理解
到達目標 : このスライドに書かれている程度のことは自在にできるようにしてエラーへの対処法を身につける. 必要なパッケージのインストールが正しくできているかどうかの自力での判定 および個別のパッケージのインストール. 作業ディレクトリの変更 3. テキストエディタで自在に入出力ファイル名の変更 ( どんなファイル名のものがどこに生成されるかという全体像の把握 ) 4. ありがちなミス のところで示しているエラーメッセージとその原因をきっちり理解
<4D F736F F D204B208C5182CC94E497A682CC8DB782CC8C9F92E BD8F6494E48A722E646F6378>
 3 群以上の比率の差の多重検定法 013 年 1 月 15 日 017 年 3 月 14 日修正 3 群以上の比率の差の多重検定法 ( 対比較 ) 分割表で表記される計数データについて群間で比率の差の検定を行う場合 全体としての統計的有意性の有無は χ 検定により判断することができるが 個々の群間の差の有意性を判定するためには多重検定法が必要となる 3 群以上の比率の差を対比較で検定する方法としては
3 群以上の比率の差の多重検定法 013 年 1 月 15 日 017 年 3 月 14 日修正 3 群以上の比率の差の多重検定法 ( 対比較 ) 分割表で表記される計数データについて群間で比率の差の検定を行う場合 全体としての統計的有意性の有無は χ 検定により判断することができるが 個々の群間の差の有意性を判定するためには多重検定法が必要となる 3 群以上の比率の差を対比較で検定する方法としては
Microsoft PowerPoint ppt
 情報科学第 07 回データ解析と統計代表値 平均 分散 度数分布表 1 本日の内容 データ解析とは 統計の基礎的な値 平均と分散 度数分布表とヒストグラム 講義のページ 第 7 回のその他の欄に 本日使用する教材があります 171025.xls というファイルがありますので ダウンロードして デスクトップに保存してください 2/45 はじめに データ解析とは この世の中には多くのデータが溢れています
情報科学第 07 回データ解析と統計代表値 平均 分散 度数分布表 1 本日の内容 データ解析とは 統計の基礎的な値 平均と分散 度数分布表とヒストグラム 講義のページ 第 7 回のその他の欄に 本日使用する教材があります 171025.xls というファイルがありますので ダウンロードして デスクトップに保存してください 2/45 はじめに データ解析とは この世の中には多くのデータが溢れています
Rでゲノム・トランスクリプトーム解析
 06.08. 版 スライド 8 までは自習 当日はスライド 9 から始める予定 スライド 3-86 は当日省略予定 講習会後に各自で復習してください 第 3 部 :NGS 解析 ( 中 ~ 上級 ) ~ トランスクリプトームアセンブリ 発現量推定 ~ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected]
06.08. 版 スライド 8 までは自習 当日はスライド 9 から始める予定 スライド 3-86 は当日省略予定 講習会後に各自で復習してください 第 3 部 :NGS 解析 ( 中 ~ 上級 ) ~ トランスクリプトームアセンブリ 発現量推定 ~ 東京大学 大学院農学生命科学研究科アグリバイオインフォマティクス教育研究プログラム門田幸二 ( かどたこうじ ) [email protected]
論文題目 腸管分化に関わるmiRNAの探索とその発現制御解析
 論文題目 腸管分化に関わる microrna の探索とその発現制御解析 氏名日野公洋 1. 序論 microrna(mirna) とは細胞内在性の 21 塩基程度の機能性 RNA のことであり 部分的相補的な塩基認識を介して標的 RNA の翻訳抑制や不安定化を引き起こすことが知られている mirna は細胞分化や増殖 ガン化やアポトーシスなどに関与していることが報告されており これら以外にも様々な細胞諸現象に関与していると考えられている
論文題目 腸管分化に関わる microrna の探索とその発現制御解析 氏名日野公洋 1. 序論 microrna(mirna) とは細胞内在性の 21 塩基程度の機能性 RNA のことであり 部分的相補的な塩基認識を介して標的 RNA の翻訳抑制や不安定化を引き起こすことが知られている mirna は細胞分化や増殖 ガン化やアポトーシスなどに関与していることが報告されており これら以外にも様々な細胞諸現象に関与していると考えられている
Microsoft Word - Stattext12.doc
 章対応のない 群間の量的データの検定. 検定手順 この章ではデータ間に 対 の対応のないつの標本から推定される母集団間の平均値や中央値の比較を行ないます 検定手法は 図. のようにまず正規に従うかどうかを調べます 但し この場合はつの群が共に正規に従うことを調べる必要があります 次に 群とも正規ならば F 検定を用いて等分散であるかどうかを調べます 等分散の場合は t 検定 等分散でない場合はウェルチ
章対応のない 群間の量的データの検定. 検定手順 この章ではデータ間に 対 の対応のないつの標本から推定される母集団間の平均値や中央値の比較を行ないます 検定手法は 図. のようにまず正規に従うかどうかを調べます 但し この場合はつの群が共に正規に従うことを調べる必要があります 次に 群とも正規ならば F 検定を用いて等分散であるかどうかを調べます 等分散の場合は t 検定 等分散でない場合はウェルチ
Microsoft PowerPoint - e-stat(OLS).pptx
.pptx") 経済統計学 ( 補足 ) 最小二乗法について 担当 : 小塚匡文 2015 年 11 月 19 日 ( 改訂版 ) 神戸大学経済学部 2015 年度後期開講授業 補足 : 最小二乗法 ( 単回帰分析 ) 1.( 単純 ) 回帰分析とは? 標本サイズTの2 変数 ( ここではXとY) のデータが存在 YをXで説明する回帰方程式を推定するための方法 Y: 被説明変数 ( または従属変数 ) X: 説明変数
経済統計学 ( 補足 ) 最小二乗法について 担当 : 小塚匡文 2015 年 11 月 19 日 ( 改訂版 ) 神戸大学経済学部 2015 年度後期開講授業 補足 : 最小二乗法 ( 単回帰分析 ) 1.( 単純 ) 回帰分析とは? 標本サイズTの2 変数 ( ここではXとY) のデータが存在 YをXで説明する回帰方程式を推定するための方法 Y: 被説明変数 ( または従属変数 ) X: 説明変数
Agilent Microarray Total Solution 5 5 RNA-Seq 60 mer DNA in situ DNA 5 2 QC 4200 TapeStation 2100 / mirna CGHCGH+SNP ChIP-on-chip 2 mirna QC
 Microarray Agilent Microarray Total Solution Agilent Microarray Total Solution 5 5 RNA-Seq 60 mer DNA in situ DNA 5 2 QC 4200 TapeStation 2100 / mirna CGHCGH+SNP ChIP-on-chip 2 mirna QC RNA / mirna total
Microarray Agilent Microarray Total Solution Agilent Microarray Total Solution 5 5 RNA-Seq 60 mer DNA in situ DNA 5 2 QC 4200 TapeStation 2100 / mirna CGHCGH+SNP ChIP-on-chip 2 mirna QC RNA / mirna total
分子系統解析における様々な問題について 田辺晶史
 分子系統解析における様々な問題について 田辺晶史 そもそもどこの配列を使うべき? そもそもどこの配列を使うべき? 置換が早すぎず遅すぎない (= 多すぎず少なすぎない ) そもそもどこの配列を使うべき? 置換が早すぎず遅すぎない (= 多すぎず少なすぎない ) 連続長は長い方が良い そもそもどこの配列を使うべき? 置換が早すぎず遅すぎない (= 多すぎず少なすぎない ) 連続長は長い方が良い 遺伝子重複が起きていない
分子系統解析における様々な問題について 田辺晶史 そもそもどこの配列を使うべき? そもそもどこの配列を使うべき? 置換が早すぎず遅すぎない (= 多すぎず少なすぎない ) そもそもどこの配列を使うべき? 置換が早すぎず遅すぎない (= 多すぎず少なすぎない ) 連続長は長い方が良い そもそもどこの配列を使うべき? 置換が早すぎず遅すぎない (= 多すぎず少なすぎない ) 連続長は長い方が良い 遺伝子重複が起きていない
基礎統計
 基礎統計 第 11 回講義資料 6.4.2 標本平均の差の標本分布 母平均の差 標本平均の差をみれば良い ただし, 母分散に依存するため場合分けをする 1 2 3 分散が既知分散が未知であるが等しい分散が未知であり等しいとは限らない 1 母分散が既知のとき が既知 標準化変量 2 母分散が未知であり, 等しいとき 分散が未知であるが, 等しいということは分かっているとき 標準化変量 自由度 の t
基礎統計 第 11 回講義資料 6.4.2 標本平均の差の標本分布 母平均の差 標本平均の差をみれば良い ただし, 母分散に依存するため場合分けをする 1 2 3 分散が既知分散が未知であるが等しい分散が未知であり等しいとは限らない 1 母分散が既知のとき が既知 標準化変量 2 母分散が未知であり, 等しいとき 分散が未知であるが, 等しいということは分かっているとき 標準化変量 自由度 の t
スライド 1
 データ解析特論重回帰分析編 2017 年 7 月 10 日 ( 月 )~ 情報エレクトロニクスコース横田孝義 1 ( 単 ) 回帰分析 単回帰分析では一つの従属変数 ( 目的変数 ) を 一つの独立変数 ( 説明変数 ) で予測する事を考える 具体的には y = a + bx という回帰直線 ( モデル ) でデータを代表させる このためにデータからこの回帰直線の切片 (a) と傾き (b) を最小
データ解析特論重回帰分析編 2017 年 7 月 10 日 ( 月 )~ 情報エレクトロニクスコース横田孝義 1 ( 単 ) 回帰分析 単回帰分析では一つの従属変数 ( 目的変数 ) を 一つの独立変数 ( 説明変数 ) で予測する事を考える 具体的には y = a + bx という回帰直線 ( モデル ) でデータを代表させる このためにデータからこの回帰直線の切片 (a) と傾き (b) を最小
カイ二乗フィット検定、パラメータの誤差
 統計的データ解析 008 008.. 林田清 ( 大阪大学大学院理学研究科 ) 問題 C (, ) ( x xˆ) ( y yˆ) σ x πσ σ y y Pabx (, ;,,, ) ˆ y σx σ y = dx exp exp πσx ただし xy ˆ ˆ はyˆ = axˆ+ bであらわされる直線モデル上の点 ( ˆ) ( ˆ ) ( ) x x y ax b y ax b Pabx (,
統計的データ解析 008 008.. 林田清 ( 大阪大学大学院理学研究科 ) 問題 C (, ) ( x xˆ) ( y yˆ) σ x πσ σ y y Pabx (, ;,,, ) ˆ y σx σ y = dx exp exp πσx ただし xy ˆ ˆ はyˆ = axˆ+ bであらわされる直線モデル上の点 ( ˆ) ( ˆ ) ( ) x x y ax b y ax b Pabx (,
Medical3
 Chapter 1 1.4.1 1 元配置分散分析と多重比較の実行 3つの治療法による測定値に有意な差が認められるかどうかを分散分析で調べます この例では 因子が1つだけ含まれるため1 元配置分散分析 one-way ANOVA の適用になります また 多重比較法 multiple comparison procedure を用いて 具体的のどの治療法の間に有意差が認められるかを検定します 1. 分析メニュー
Chapter 1 1.4.1 1 元配置分散分析と多重比較の実行 3つの治療法による測定値に有意な差が認められるかどうかを分散分析で調べます この例では 因子が1つだけ含まれるため1 元配置分散分析 one-way ANOVA の適用になります また 多重比較法 multiple comparison procedure を用いて 具体的のどの治療法の間に有意差が認められるかを検定します 1. 分析メニュー
ThermoFisher
 Thermo Fisher Connect Relative Quantification 操作簡易資料 http://www.thermofisher.com/cloud 使用には事前登録が必要になります 画面は予告なく変わることがあります The world leader in serving science Thermo Fisher Connect とは? キャピラリシーケンサ リアルタイム
Thermo Fisher Connect Relative Quantification 操作簡易資料 http://www.thermofisher.com/cloud 使用には事前登録が必要になります 画面は予告なく変わることがあります The world leader in serving science Thermo Fisher Connect とは? キャピラリシーケンサ リアルタイム
NGS_KAPA RNA HyperPrep Kit
 シークエンシングワークフロー ライブラリー調製 サンプル 調製 末端修復 エンドポイントライブラリー増幅 A-TAILING アダプター ライゲーション サイズセレクション & サイズ確認 または リアルタイムライブラリー増幅 ライブラリー 定量 クラスター 増幅 KAPA RNA HyperPrep Kit illumina社用ライブラリー調製キット KAPA RNA HyperPrep Kit
シークエンシングワークフロー ライブラリー調製 サンプル 調製 末端修復 エンドポイントライブラリー増幅 A-TAILING アダプター ライゲーション サイズセレクション & サイズ確認 または リアルタイムライブラリー増幅 ライブラリー 定量 クラスター 増幅 KAPA RNA HyperPrep Kit illumina社用ライブラリー調製キット KAPA RNA HyperPrep Kit
第 10 回シーケンス講習会 RNA-seq library 調製法の特徴と選び方 理化学研究所 (RIKEN) ライフサイエンス技術基盤研究センター (CLST) 機能性ゲノム解析部門 (DGT) ゲノムネットワーク解析支援施設 (GeNAS) 野間将平
 ライフサイエンス技術基盤研究センター (CLST) 機能性ゲノム解析部門 (DGT) ゲノムネットワーク解析支援施設 (GeNAS) 野間将平") 第 10 回シーケンス講習会 RNA-seq library 調製法の特徴と選び方 理化学研究所 (RIKEN) ライフサイエンス技術基盤研究センター (CLST) 機能性ゲノム解析部門 (DGT) ゲノムネットワーク解析支援施設 (GeNAS) 野間将平 l シーケンスをする目的は? 概略 l よいシーケンスライブラリーとは? RNA-seq ライブラリーのムリ ムダ ムラ l いろいろな RNA-seq
第 10 回シーケンス講習会 RNA-seq library 調製法の特徴と選び方 理化学研究所 (RIKEN) ライフサイエンス技術基盤研究センター (CLST) 機能性ゲノム解析部門 (DGT) ゲノムネットワーク解析支援施設 (GeNAS) 野間将平 l シーケンスをする目的は? 概略 l よいシーケンスライブラリーとは? RNA-seq ライブラリーのムリ ムダ ムラ l いろいろな RNA-seq
CAEシミュレーションツールを用いた統計の基礎教育 | (株)日科技研
日科技研") CAE シミュレーションツール を用いた統計の基礎教育 ( 株 ) 日本科学技術研修所数理事業部 1 現在の統計教育の課題 2009 年から統計教育が中等 高等教育の必須科目となり, 大学でも問題解決ができるような人材 ( 学生 ) を育てたい. 大学ではコンピューター ( 統計ソフトの利用 ) を重視した教育をより積極的におこなうのと同時に, 理論面もきちんと教育すべきである. ( 報告 数理科学分野における統計科学教育
CAE シミュレーションツール を用いた統計の基礎教育 ( 株 ) 日本科学技術研修所数理事業部 1 現在の統計教育の課題 2009 年から統計教育が中等 高等教育の必須科目となり, 大学でも問題解決ができるような人材 ( 学生 ) を育てたい. 大学ではコンピューター ( 統計ソフトの利用 ) を重視した教育をより積極的におこなうのと同時に, 理論面もきちんと教育すべきである. ( 報告 数理科学分野における統計科学教育
Microsoft PowerPoint - Ion Reporter?ソフトウェアを用いた変異解析4.6.pptx
 Ion Reporter ソフトウェアデモンストレーション Ion AmpliSeq Comprehensive Cancer Panel を用いたがん部および非がん部の体細胞変異比較解析 1 Ion Torrent システムを用いた実験例 Ion AmpliSeq Comprehensive Cancer Panel を 2 サンプル実施 ランレポート ランレポート サンプル 1 サンプル 2 2
Ion Reporter ソフトウェアデモンストレーション Ion AmpliSeq Comprehensive Cancer Panel を用いたがん部および非がん部の体細胞変異比較解析 1 Ion Torrent システムを用いた実験例 Ion AmpliSeq Comprehensive Cancer Panel を 2 サンプル実施 ランレポート ランレポート サンプル 1 サンプル 2 2
計算機生命科学の基礎II_
 Ⅱ 1.4 [email protected] 812-8582 3-1-1 8 806 http://www.cell-innovator.com BioGPS Connectivity Map The Cancer Genome Atlas (TCGA); cbioportal GO DAVID, GSEA WCGNA BioGPS http://biogps.org/
Ⅱ 1.4 [email protected] 812-8582 3-1-1 8 806 http://www.cell-innovator.com BioGPS Connectivity Map The Cancer Genome Atlas (TCGA); cbioportal GO DAVID, GSEA WCGNA BioGPS http://biogps.org/
スライド 1
 データ解析特論第 10 回 ( 全 15 回 ) 2012 年 12 月 11 日 ( 火 ) 情報エレクトロニクス専攻横田孝義 1 終了 11/13 11/20 重回帰分析をしばらくやります 12/4 12/11 12/18 2 前回から回帰分析について学習しています 3 ( 単 ) 回帰分析 単回帰分析では一つの従属変数 ( 目的変数 ) を 一つの独立変数 ( 説明変数 ) で予測する事を考える
データ解析特論第 10 回 ( 全 15 回 ) 2012 年 12 月 11 日 ( 火 ) 情報エレクトロニクス専攻横田孝義 1 終了 11/13 11/20 重回帰分析をしばらくやります 12/4 12/11 12/18 2 前回から回帰分析について学習しています 3 ( 単 ) 回帰分析 単回帰分析では一つの従属変数 ( 目的変数 ) を 一つの独立変数 ( 説明変数 ) で予測する事を考える
2016_RNAseq解析_修正版
 平成 28 年度 NGS ハンズオン講習会 RNA-seq 解析 2016 年 7 27 本講義にあたって n 代表的な解析の流れを紹介します 論 でよく使 されているツールを使 します n コマンドを沢 実 します タイプミスが 配な は コマンド例がありますのでコピーして実 してください 実 が遅れてもあせらずに 課題や休憩の間に追い付いてください Amelieff Corporation All
平成 28 年度 NGS ハンズオン講習会 RNA-seq 解析 2016 年 7 27 本講義にあたって n 代表的な解析の流れを紹介します 論 でよく使 されているツールを使 します n コマンドを沢 実 します タイプミスが 配な は コマンド例がありますのでコピーして実 してください 実 が遅れてもあせらずに 課題や休憩の間に追い付いてください Amelieff Corporation All
Python-statistics5 Python で統計学を学ぶ (5) この内容は山田 杉澤 村井 (2008) R によるやさしい統計学 (
 この内容は山田 杉澤 村井 (2008) R によるやさしい統計学 (") http://localhost:8888/notebooks/... Python で統計学を学ぶ (5) この内容は山田 杉澤 村井 (2008) R によるやさしい統計学 (http://shop.ohmsha.co.jp/shop /shopdetail.html?brandcode=000000001781&search=978-4-274-06710-5&sort=) を参考にしています
http://localhost:8888/notebooks/... Python で統計学を学ぶ (5) この内容は山田 杉澤 村井 (2008) R によるやさしい統計学 (http://shop.ohmsha.co.jp/shop /shopdetail.html?brandcode=000000001781&search=978-4-274-06710-5&sort=) を参考にしています
分子系統解析における様々な問題について 田辺晶史
 分子系統解析における様々な問題について 田辺晶史 そもそもどこの配列を使うべき? そもそもどこの配列を使うべき? 置換が早すぎず遅すぎない (= 多すぎず少なすぎない ) そもそもどこの配列を使うべき? 置換が早すぎず遅すぎない (= 多すぎず少なすぎない ) 連続長は長い方が良い そもそもどこの配列を使うべき? 置換が早すぎず遅すぎない (= 多すぎず少なすぎない ) 連続長は長い方が良い 遺伝子重複が起きていない
分子系統解析における様々な問題について 田辺晶史 そもそもどこの配列を使うべき? そもそもどこの配列を使うべき? 置換が早すぎず遅すぎない (= 多すぎず少なすぎない ) そもそもどこの配列を使うべき? 置換が早すぎず遅すぎない (= 多すぎず少なすぎない ) 連続長は長い方が良い そもそもどこの配列を使うべき? 置換が早すぎず遅すぎない (= 多すぎず少なすぎない ) 連続長は長い方が良い 遺伝子重複が起きていない
数量的アプローチ 年 6 月 11 日 イントロダクション データ分析をマスターする 12 のレッスン ウェブサポートページ ( 有斐閣 ) 水落研究室 R http:
 水落研究室 R http:") イントロダクション データ分析をマスターする 12 のレッスン ウェブサポートページ ( 有斐閣 ) http://yuhikaku-nibu.txt-nifty.com/blog/2017/09/22103.html 水落研究室 R http://depts.nanzan-u.ac.jp/ugrad/ps/mizuochi/r.html 1 この授業では統計ソフト R を使って分析を行います データを扱うソフトとして
イントロダクション データ分析をマスターする 12 のレッスン ウェブサポートページ ( 有斐閣 ) http://yuhikaku-nibu.txt-nifty.com/blog/2017/09/22103.html 水落研究室 R http://depts.nanzan-u.ac.jp/ugrad/ps/mizuochi/r.html 1 この授業では統計ソフト R を使って分析を行います データを扱うソフトとして
Fortran 勉強会 第 5 回 辻野智紀
 Fortran 勉強会 第 5 回 辻野智紀 今回のお品書き サブルーチンの分割コンパイル ライブラリ 静的ライブラリ 動的ライブラリ モジュール その前に 以下の URL から STPK ライブラリをインストールしておいて下さい. http://www.gfd-dennou.org/library/davis/stpk 前回参加された方はインストール済みのはず. サブルーチンの分割コンパイル サブルーチンの独立化
Fortran 勉強会 第 5 回 辻野智紀 今回のお品書き サブルーチンの分割コンパイル ライブラリ 静的ライブラリ 動的ライブラリ モジュール その前に 以下の URL から STPK ライブラリをインストールしておいて下さい. http://www.gfd-dennou.org/library/davis/stpk 前回参加された方はインストール済みのはず. サブルーチンの分割コンパイル サブルーチンの独立化
Microsoft PowerPoint - 資料04 重回帰分析.ppt
 04. 重回帰分析 京都大学 加納学 Division of Process Control & Process Sstems Engineering Department of Chemical Engineering, Koto Universit [email protected] http://www-pse.cheme.koto-u.ac.jp/~kano/ Outline
04. 重回帰分析 京都大学 加納学 Division of Process Control & Process Sstems Engineering Department of Chemical Engineering, Koto Universit [email protected] http://www-pse.cheme.koto-u.ac.jp/~kano/ Outline